Housed in a state-of-the-art building to the east of central Oxford, UK, is the Kennedy Institute of Rheumatology. The centre forms part of the University of Oxford’s Nuffield Department of Orthopaedics, Rheumatology, and Musculoskeletal Sciences (NDORMS), but this association with Oxford is only a recent development in the five decade-long history of this institute.
Preference: Rheumatology
1067
[Perspectives] Marc Feldmann: a trailblazer of modern immunology
Marc Feldmann, Emeritus Professor at the Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences at the University of Oxford, UK, retired from directing the Kennedy Institute of Rheumatology 2 years ago, and is enjoying a new lease of life, dividing his time between Oxford and New York City’s Mount Sinai Medical School. Research for Feldmann now means a focus on translational science with potential clinical impact, mainly done by mentoring younger researchers.
Osteoporosis treatment: a missed opportunity
Minimal trauma fractures remain a major cause of morbidity in Australia, affecting one in two women and one in four men over the age of 60 years.1 Mortality is increased after all minimal trauma fractures, even after minor fractures.2 Hip fractures are particularly devastating, leading to decreased quality of life, increased mortality and loss of functional independence.3
Defining osteoporosis
Bone mineral density (BMD) is expressed in relation to either “young normal” adults of the same sex (T score) or to the expected BMD for the patient’s age and sex (Z score). Osteoporosis is defined as a T score ≤ 2.5 SDs below that of a “young normal” adult, with fracture risk increasing twofold to threefold for each SD decrease in BMD.4,5 A BMD Z score less than −2 indicates that BMD is below the normal range for age and sex, and warrants a more intensive search for secondary causes. Importantly, osteoporosis is also diagnosed after a minimal trauma fracture, irrespective of the patient’s T score.
Absolute fracture risk
Treatment for osteoporosis is recommended for patients with a high absolute fracture risk. This includes older Australians (post-menopausal women and men aged over 60 years) with T scores ≤ −2.5 at the lumbar spine, femoral neck or total hip, and patients with a history of a minimal trauma fracture.6 There is a major gap between evidence and treatment in secondary fracture prevention, with fewer than 20% of patients presenting with a minimal trauma fracture being treated or investigated for osteoporosis.7,8 However, it is important that patients with a low fracture risk, including younger women without clinical risk factors and T scores ≤ −2.5 at “non-main-sites” (eg, lateral lumbar spine or Ward’s triangle in the hip) are not treated.9 Absolute fracture risk calculators incorporate osteoporosis risk factors with BMD to stratify fracture probability.10 It is therefore important for clinicians to assess absolute fracture risk. Two of several absolute fracture risk calculators are commonly used to aid clinicians in this regard: the Garvan Fracture Risk Calculator11 and the Fracture Risk Assessment Tool (FRAX) developed by the World Health Organization.
The Garvan Fracture Risk Calculator estimates absolute fracture risk over 5 and 10 years (http://www.garvan.org.au/bone-fracture-risk/). It may be used in men and women aged over 50 years, and incorporates age, sex, BMD at the spine or femoral neck, falls and fracture history. A potential limitation of this tool is that it does not include other clinical risk factors. The country-specific FRAX tool calculates the 10-year probability of hip fracture and major osteoporotic fracture in patients aged 40–90 years. It incorporates femoral neck BMD with ten clinical risk factors. Limitations include underestimation of fracture risk in patients with multiple minimal trauma fractures, an inability to adjust the risk for dose-dependent exposure, a lack of validation for use with BMD of the spine, and exclusion of falls.
Role of fracture risk calculators in 2016
The role of absolute fracture risk calculators in clinical practice is evolving. In addition to their individual limitations, there is a lack of evidence that their use leads to effective targeting of drug therapy to those deemed to be at high risk of fracture,12 and prospective studies are needed. In particular, country-specific intervention thresholds based on absolute fracture risk need to be validated clinically. However, fracture risk calculators are useful for identifying patients with low fracture risk who do not require treatment.
Special patient groups
Limited evidence-based guidance is available for treating osteoporosis in several groups, including patients with post-transplantation osteoporosis, type 1 diabetes mellitus, chronic kidney disease (creatinine clearance < 30 mL/minute), neurological, respiratory and haematological diseases, and young adults and pregnant women. Such patients require individualised management.
Osteoporosis prevention using non-pharmacological therapies
Lifestyle approaches (adequate dietary calcium intake, optimal vitamin D status, participation in resistance exercise, smoking cessation, avoidance of excessive alcohol, falls prevention) act as a framework for improving musculoskeletal health at a population-based level.6,13–16
Calcium and vitamin D
The current Australian recommended daily intake (RDI) of calcium is 1300 mg per day for women aged over 51 years, 1000 mg per day for men aged 51–70 years and 1300 mg per day for men aged over 70 years.17 Adverse effects of calcium supplementation include gastrointestinal bloating, constipation,18 and renal calculi.19 There is controversy about the efficacy of calcium in preventing osteoporotic fractures.6,19,20 Further work is required with studies powered to investigate cardiac outcomes in men and women receiving calcium supplementation to meet current RDIs. Higher dietary calcium intake is also associated with reductions in mortality, cardiovascular events and strokes.21 Dietary sources of calcium are the preferred sources. Calcium supplementation should be limited to 500–600 mg per day, and used only by those who cannot achieve the RDI with dietary calcium.15
The main source of vitamin D is through exposure to sunlight. Institutionalised or housebound older people are at particularly high risk of vitamin D deficiency. Inadequate vitamin D status is defined as a serum 25-hydroxyvitamin D (25(OH)D) level < 50 nmol/L in late winter/early spring; in older individuals such inadequate vitamin D levels are associated with muscular weakness and decreased physical performance.22 Increased falls and fractures occur at 25(OH)D levels < 25–30 nmol/L.23,24 Adults aged 50–70 years and those over 70 years require at least 600 IU to 800 IU of vitamin D3 daily, with larger daily doses required to treat vitamin D deficiency.25
Exercise
Community-based high speed, power training, multimodal exercise programs increase BMD and muscle strength, with a trend to falls reduction.26 Thus, exercise is recommended both to maintain bone health and reduce falls. It should be individualised to the patient’s needs and abilities, increasing progressively as tolerated by the degree of osteoporosis-related disability.
Falls prevention
Falls are the precipitating factor in nearly 90% of all appendicular fractures, including hip fractures,3 and reducing falls risk is critical in managing osteoporosis. Reducing the use of benzodiazepines, neuroleptic agents and antidepressants reduces the risk of falls,27 and, among women aged 75 or more years, muscle strengthening and balance exercises reduce the risk of both falls and injuries.28
Antiresorptive therapy for osteoporosis
Post-menopausal osteoporosis results from an imbalance in bone remodelling, such that bone resorption exceeds bone formation. Antiresorptive drugs decrease the number, activity and lifespan of osteoclasts,29 preserving or increasing bone mass with a resulting reduction in vertebral, non-vertebral and hip fractures. These drugs include bisphosphonates (oral or intravenous),30–35 oestrogen36,37 and selective oestrogen receptor-modulating drugs,38 strontium ranelate and denosumab, a human monoclonal antibody against receptor activator of nuclear factor κB-ligand (RANKL).39
Antiresorptive treatments for osteoporosis are approved for reimbursement on the Pharmaceutical Benefits Scheme (PBS) for men and post-menopausal women following a minimal trauma fracture, as well as for those at high risk of fracture, on the basis of age (> 70 years) and low BMD (T score < −2.5 or −3.0). Bisphosphonates are also approved for premenopausal women who have had a minimal trauma fracture. In patients at high risk of fracture, osteoporosis therapy reduces the risk of vertebral fractures by 40–70%, non-vertebral fractures by about 25%, and hip fractures by 40–50%.30–40
Bisphosphonates
Mechanism of action and efficacy. Bisphosphonates are stable analogues of pyrophosphate. They bind avidly to hydroxyapatite crystals on bone and are then released slowly at sites of active bone remodelling in the skeleton, leading to recirculation of bisphosphonates. The terminal half-lives of bisphosphonates differ; for alendronate it is more than 10 years,41 while for risedronate it is about 3 months.42
Alendronate prevents minimal trauma fractures. Therapy with alendronate reduces vertebral fracture risk by 48% compared with placebo. Similar reductions in the risk of hip and wrist fractures were seen in women treated with alendronate who had low BMD and prevalent vertebral fractures.33,34,43 A randomised, double-blind, placebo-controlled trial of post-menopausal women assigned to risedronate therapy or placebo for 3 years showed vertebral and non-vertebral fracture risks were respectively reduced by 41% and 39% by risedronate.35 Three years of treatment with zoledronic acid in women with post-menopausal osteoporosis reduced the risk of morphometric vertebral fracture by 70% compared with placebo, and reduced the risk of non-vertebral and hip fracture by 25% and 41% respectively.30
Adverse effects. The main potential adverse effects of oral bisphosphonates are gastrointestinal (including reflux, oesophagitis, gastritis and diarrhoea). Oral bisphosphonates should not be given to patients with active upper gastrointestinal disease, dysphagia or achlasia. Intravenous bisphosphonates are associated with an acute phase reaction (fever, flu-like symptoms, myalgias, headache and arthralgia) in about a third of patients, typically within 24–72 hours of receiving their first infusion of zoledronic acid, but is reduced significantly on subsequent infusions.30 Treatment with antipyretic agents, including paracetamol, improves these symptoms. Treatment with bisphosphonates may also lower serum calcium concentrations, but this is uncommon in the absence of vitamin D deficiency.44,45 Bisphosphonates are not recommended for use in patients with creatinine clearance below 30–35 mL/min.
Less common adverse effects associated with long term bisphosphonate therapy include osteonecrosis of the jaw (ONJ) and atypical femoral fracture (AFF). Overemphasis of these uncommon adverse effects by patients has led to declining osteoporosis treatment rates.46
Jaw osteonecrosis. ONJ is said to occur when there is an area of exposed bone in the maxillofacial region that does not heal within 8 weeks after being identified by a health care provider, in a patient who was receiving or had been exposed to a bisphosphonate and did not have radiation therapy to the craniofacial region.47 Risk factors for ONJ include intravenous bisphosphonate therapy for malignancy, chemotherapeutic agents, duration of exposure to bisphosphonates, dental extractions, dental implants, poorly fitting dentures, glucocorticoid therapy, smoking, diabetes and periodontal disease.48,49 The risk of ONJ is about 1 in 10 000 to 1 in 100 000 patient-years in patients taking oral bisphosphonates for osteoporosis.47 Given the prolonged half-life of bisphosphonates, temporary withdrawal of treatment before extractions is unlikely to have a significant benefit and is therefore not recommended.50
Atypical femur fractures. Clinical trial data clearly support the beneficial effect of bisphosphonates in preventing minimal trauma fractures. However, oversuppression of bone remodelling may allow microdamage to accumulate, leading to increased bone fragility.51 Cases of AFF and severely suppressed bone remodelling after prolonged bisphosphonate therapy52 have prompted further research and recent guideline development.53 However, this finding is not universal. AFFs occur in the subtrochanteric region or diaphysis of the femur and have unique radiological features, including a predominantly transverse fracture line, periosteal callus formation and minimal comminution, as shown in Box 1.53 AFFs have been reported in patients taking bisphosphonates and denosumab, but about 7% of cases occur without exposure to either drug. AFFs appear to be more common in patients who have been exposed to long term bisphosphonate therapy, with a higher risk (113 per 100 000 person-years) in patients who receive more than 7–8 years of therapy.53 Although many research questions remain unanswered, including aetiology, optimal screening and management of these fractures, the risk of a subsequent AFF is reduced from 12 months after cessation of bisphosphonate treatment.
Duration of therapy. Concerns about the small but increased risk of adverse events after long term treatment with bisphosphonates (Box 2) have led to the development of guidelines on the optimal duration of therapy.54 For patients at high risk of fracture, bisphosphonate treatment for up to 10 years (oral) or 6 years (intravenous) is recommended. For women who are not at high risk of fracture after 3 years of intravenous or 5 years of oral bisphosphonate treatment, a drug holiday of 2–3 years may be considered (Box 3). However, it is critical to understand that “holiday” does mean “retirement”, and those patients should continue to have BMD monitoring after 2–3 years.
Hormone replacement therapy
Hormone replacement therapy (HRT) is effective in preventing and treating post-menopausal osteoporosis. Benefits need to be balanced against thromboembolic and vascular risk, breast cancer risk (for oestrogen plus progesterone), and duration of therapy. HRT is most suitable for recently menopausal woman (up until age 59 years), particularly for those with menopausal symptoms. In women with an early or premature menopause, HRT should be continued until the average age of menopause onset (about 51 years), or longer in the setting of a low BMD. Oral or transdermal oestrogen therapy (in women who have had a hysterectomy) and combined oestrogen and progesterone therapy preserve BMD,55 and were also shown to reduce the risk of hip, vertebral and total fractures compared with placebo in the Women’s Health Initiative (WHI).37,56
In the initial WHI analysis, combined oral oestrogen and progesterone therapy for 5.6 years in post-menopausal women aged 50–79 years (who were generally older than women who used HRT for control of menopausal symptoms), many of whom had cardiovascular risk factors, was shown to increase the risk of breast cancer, stroke and thromboembolic events.57 However, subsequent reanalysis of WHI data has established the efficacy and safety of HRT in younger women up until 10 years after menopause, or the age of 59 years, when the benefits of treatment outweigh the risks. In women with a history of hysterectomy, oral oestrogen therapy alone has a better benefit–risk profile, with no increases in rates of breast cancer or coronary heart disease.56
Women commencing HRT should be fully informed about its benefits and risks. Cardiovascular risk is not increased when therapy is initiated within 10 years of menopause,58,59 but the risk of stroke is elevated regardless of time since menopause. It is also recommended that doctors discuss smoking cessation, blood pressure control and treatment of dyslipidaemia with women commencing HRT.
Selective oestrogen receptor modulator (SERM) drugs
The SERM raloxifene has beneficial oestrogen-like effects on bone, but has oestrogen antagonist activity on breast and endometrium. Treatment with raloxifene for 3 years reduced vertebral fractures by 30–50% compared with placebo in post-menopausal women.38 However, there was no reduction in non-vertebral fractures. Consequently, raloxifene is useful in post-menopausal women with spinal osteoporosis, particularly those with an increased risk of breast cancer. Raloxifene therapy is also associated with a 72% reduction in the risk of invasive breast cancer.60 Raloxifene may exacerbate hot flushes, and women receiving raloxifene have a greater than threefold increased incidence of thromboembolic disease, comparable with those receiving HRT.36,56 Raloxifene therapy is also associated with an increased risk of stroke,61 particularly in current smokers.
Denosumab
Denosumab is a human monoclonal antibody with specificity for RANKL, which stimulates the development and activity of osteoclasts. Denosumab mimics the endogenous inhibitor of RANKL, osteoprotegerin, and is given as a 60 mg subcutaneous injection once every 6 months. Denosumab reduces new clinical vertebral fractures by 68%, with a 40% reduction in hip fracture and a 20% reduction in non-vertebral fractures compared with placebo over 3 years.39,62
The adverse effects of denosumab include small increases in the risks of eczema, cellulitis and flatulence.39 Hypocalcaemia, particularly in patients with abnormal renal function, has also been reported,63 and denosumab is contraindicated in patients with hypocalcaemia. Jaw osteonecrosis has been reported in patients receiving denosumab for osteoporosis, as have AFFs.64,65
Strontium ranelate
Strontium ranelate increases bone formation markers and reduces bone resorption markers, but is predominantly antiresorptive, as increases in the rate of bone formation have not been demonstrated.66 Strontium ranelate significantly reduces the risk of vertebral and non-vertebral fractures.67–69 The most frequent adverse effects associated with strontium ranelate are nausea, diarrhoea, headache, dermatitis and eczema.67,68 Cases of a rare hypersensitivity syndrome (drug reaction, eosinophilia and systemic symptoms [DRESS]) have been reported, and strontium ranelate should be discontinued if a rash develops. Strontium ranelate treatment was associated with an increased incidence of venous thromboembolism70 and, more recently, with a small increase in absolute risk of acute myocardial infarction. Strontium ranelate is contraindicated in patients with uncontrolled hypertension and/or a current or past history of ischaemic heart disease, peripheral arterial disease and/or cerebrovascular disease.71 This drug is now a second-line treatment for osteoporosis, only used when other medications for osteoporosis are unsuitable, in the absence of contraindications.
Anabolic therapy for osteoporosis
Teriparatide
Teriparatide increases osteoblast recruitment and activity to stimulate bone formation.40 In contrast to antiresorptive agents, which preserve bone microarchitecture and inhibit bone loss, teriparatide (recombinant human parathyroid hormone [1–34]) stimulates new bone formation and improves bone microarchitecture. Teriparatide reduced the risk of new vertebral fractures by 65% in women with osteoporosis who have had one or more baseline fractures40 and also reduced new or worsening back pain. Non-vertebral fractures are also reduced by 53% by teriparatide, but studies have been underpowered to detect reductions in the rate of hip fracture. Side effects include headache (8%), nausea (8%), dizziness and injection-site reactions. Transient hypercalcaemia (serum calcium level, > 2.60 mmol/L) after dosing also occurred in 3–11% of patients receiving teriparatide.
Teriparatide has a black box warning concerning an increased incidence of osteosarcoma in rats that were exposed to 3 and 60 times the normal human exposure over a significant portion of their lives. Teriparatide is therefore contraindicated in patients who may be at increased risk of osteosarcoma, including those with a prior history of skeletal irradiation, Paget’s disease of bone, an unexplained elevation in bone-specific alkaline phosphatase, bone disorders other than osteoporosis, and in adolescents and children.
In Australia, the maximum lifetime duration of teriparatide therapy is 18 months. However, the antifracture benefit increases the longer the patient remains on treatment, with non-vertebral fractures being reduced for up to 2 years of treatment compared with the first 6 months of treatment, and for up to 2 years following cessation of treatment.72 In addition, increases in the rates of trabecular and cortical bone formation continue for up to 2 years of treatment, refuting the outmoded concept of a limited “anabolic window” of action for this drug.73 Importantly, following teriparatide therapy, the accrued benefits will be lost if antiresorptive therapy is not immediately instituted. Teriparatide reimbursement through the PBS is restricted to patients who have had two minimal trauma fractures and who have a fracture after at least a year of antiresorptive therapy, and who have a BMD T score below −3. However, the rate of teriparatide use in Australia is among the lowest in the world (David Kendler, University of British Columbia, Canada, personal communication).
Future directions
Three new anti-osteoporosis drugs are in clinical development.
“Selective” antiresorptive drugs
A novel “selective” antiresorptive drug, odanacatib, is a cathepsin K inhibitor that has the advantage of not suppressing bone formation, as do traditional or “non-selective” antiresorptive drugs. Clinical trial data in the largest ever osteoporosis trial, published in abstract form, show that odanacatib, given as a weekly tablet, reduces vertebral, non-vertebral and hip fractures with risk reductions similar to those seen with bisphosphonates. Adverse events were reported and include atypical femur fractures, morphea and adjudicated cerebrovascular events.74 The benefit–risk profile of this drug is currently being clarified.
Anabolic drugs
The two other new drugs are anabolic agents. Abaloparatide, an analogue of parathyroid hormone-related protein (1–34), selectively acts on the type 1 parathyroid hormone receptor to stimulate bone formation. It is given as a daily injection.75 It reduces vertebral and non-vertebral fractures, but data for hip fracture are lacking.76 Abaloparatide reduced major osteoporotic fractures by 67% compared with placebo.77 Abaloparatide will also have a black box warning about osteogenic sarcoma in rats. The final drug, romosozumab, is a monoclonal antibody that targets an inhibitor of bone formation, sclerostin, and is given as 2-monthly injections for 12 months. Trial data comparing reductions in fractures with placebo are awaited, and a head-to-head trial comparing the antifracture efficacy of romosozumab with alendronate is ongoing.
Conclusion
Osteoporosis treatment represents a missed opportunity for medical practitioners. Despite a growing number of effective therapies, where the benefits far outweigh the risks, only a minority of patients presenting to the health care system with minimal trauma fractures are being either investigated or treated for osteoporosis.
The time to close this gap between evidence and treatment is long overdue and will require systems-based approaches supported by both the federal and state governments. One such approach is fracture liaison services, which have proven efficacy in cost-effectively reducing the burden of fractures caused by osteoporosis, and are increasingly being implemented internationally. General practitioners also need to take up the challenge imposed by osteoporosis and become the champions of change, working with the support of specialists and government to reduce the burden of fractures caused by osteoporosis in Australia.
Box 1 –
Bilateral atypical femoral fractures in an older woman after bisphosphonate therapy for 9 years*
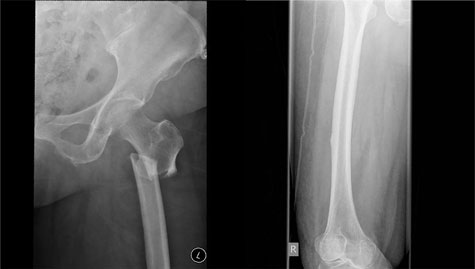
* Note the characteristic findings of a predominantly transverse fracture line, periosteal callus formation and minimal comminution on the left, and the periosteal reaction on the lateral cortex on the right femur, indicating an early stress fracture.
Box 2 –
Balancing benefits and risks of bisphosphonate therapy with other lifetime risks*
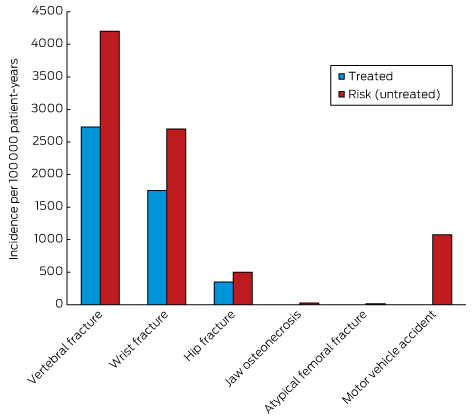
* Adapted from Adler, et al.54
Box 3 –
Approach to the management of post-menopausal women on long term bisphosphonates therapy for osteoporosis*
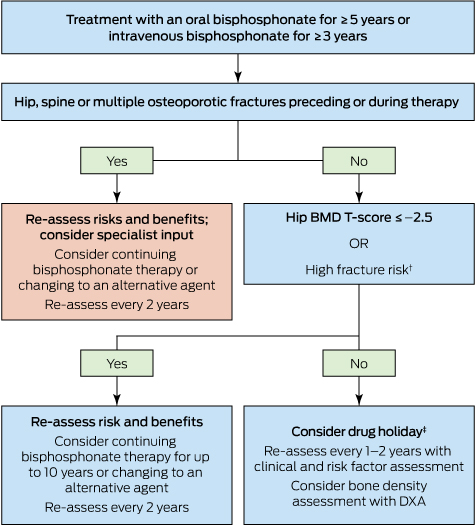
DXA = dual-energy x-ray absorptiometry. * Adapted from Adler, et al.54 † Includes age > 70 years; clinical risk factors for fracture and osteoporosis; fracture risk score on fracture risk calculation tools above the Australian treatment threshold. ‡ Cessation of treatment for 2–3 years.
Cardiac tamponade in undiagnosed systemic lupus erythematosus
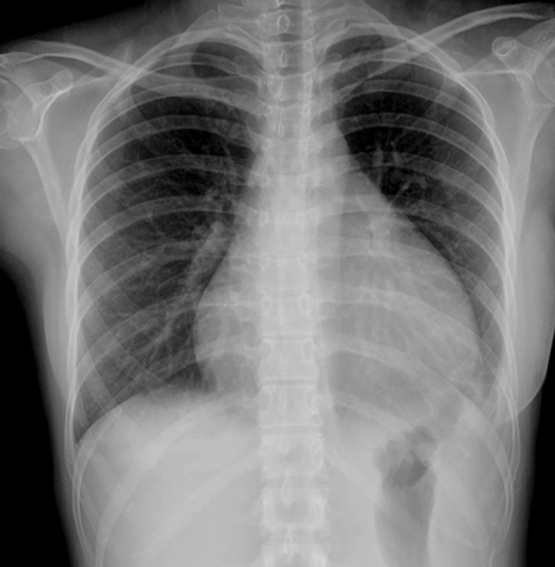
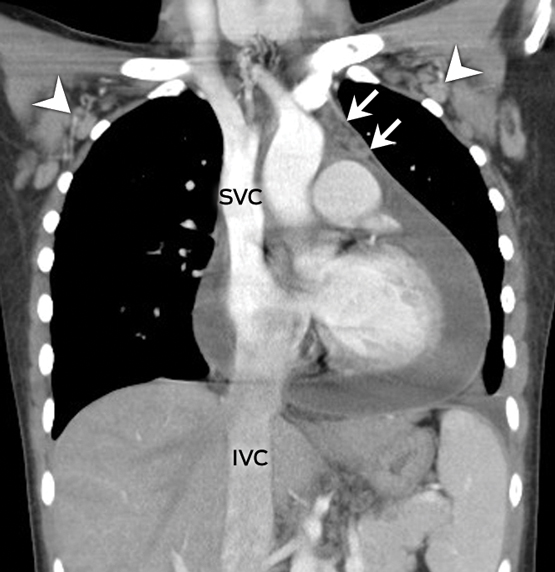
A 22-year-old woman presented with a 3-day history of fever, retrosternal chest pain and exertional dyspnoea. Her heart rate was 130 bpm with a blood pressure level of 109/68 mmHg. Physical examination suggested tamponade: distended jugular veins, pulsus paradoxus and muffled heart tones. The chest radiography was notable for the characteristic water-bottle sign (Figure, A).1 Contrast-enhanced chest computed tomography demonstrated a massive pericardial effusion (Figure, B) associated with venous engorgement of the superior and inferior vena cava (SVC, IVC), prevascular space (arrows), and bilateral axillary veins (arrowheads). An emergency thoracoscopic pericardial window was performed and 620 mL of bloody fluid was drained.
The presence of anti-nuclear, anti-double-stranded DNA, anti-Smith antibodies and hypocomplementaemia supported the diagnosis of systemic lupus erythematosus.2 The patient recovered after 1 week of intravenous methylprednisolone pulse therapy. At an 8-month follow-up, there have been no recurrences.
[Comment] Beyond methotrexate monotherapy for early rheumatoid arthritis
Findings from the U-Act-Early strategy trial in patients with early rheumatoid arthritis by Johannes Bijlsma and colleagues1 in The Lancet suggest that current standard care initiating monotherapy with the conventional synthetic disease-modifying antirheumatic drug (DMARD) methotrexate is suboptimal. The trial investigators enrolled 317 patients from 21 Dutch rheumatology outpatient departments and randomly assigned them to start tocilizumab (an interleukin-6 receptor-blocking monoclonal antibody) plus methotrexate, or tocilizumab, or methotrexate.
Identifying low-value care: the Royal Australasian College of Physicians’ EVOLVE initiative
Challenges and lessons arising from early adoption of a new approach towards determining what is good clinical practice
In March 2015, 41 medical specialties of the Royal Australasian College of Physicians (RACP) came together as part of the College’s EVOLVE initiative. The main aim of EVOLVE is to drive safer, higher-quality patient care through identifying and reducing low-value medical practices.1 In EVOLVE, “low-value” practices are defined as tests, procedures or interventions that are overused, inappropriate or of limited effectiveness (and, in extreme cases, potentially harmful). The name of the initiative reflects the dynamic and evolving nature of evidence-based medicine. EVOLVE is modelled on the Choosing Wisely initiative in the United States and similar initiatives underway in Canada, Italy and the United Kingdom.2
In EVOLVE’s first year, more than 20 specialties have completed or commenced work on lists of “top-five” low-value clinical practices in their respective fields. Here, we examine the approaches of three early adopter EVOLVE specialties — geriatric medicine, palliative medicine and rheumatology. We also share insights that have arisen so far that are relevant to the Medicare Benefits Schedule (MBS) Review Taskforce.
The EVOLVE approach
EVOLVE recognises the breadth of physicians’ practice, uniting specialties through their commitment to reducing low-value care. It is a partnership between specialty societies and the RACP. EVOLVE is clinician-led, with each specialty responsible for developing lists, engaging with its members and providing feedback to the RACP on systemic barriers to adoption of each list’s recommendations. The RACP is the umbrella body, developing common frameworks and a robust methodology, coordinating across and between specialties, connecting EVOLVE with associated initiatives such as Choosing Wisely Australia, and communicating about and advocating for high-value care.
To avoid the early mistakes of Choosing Wisely in the US, where some participating specialties identified “low-impact” practices on their lists and singled out clinical practices performed by other specialties,3 EVOLVE’s participating specialties agreed to robust principles and methods. These included:
-
Practices under consideration by each specialty should be “within or significantly impact their domain of practice”. This can be interpreted as including practices involving shared decision making with other health care specialties and those that are the subject of referral to and from other specialties. Specialties also have broad discretion to consider practices that they consider can “make a difference” in reducing low-value care (eg, rheumatologists and geriatric medicine specialists examined practices that affected people with conditions they commonly treated).
-
Practices under consideration should be either growing in use or currently commonly used. Some specialties interpreted “commonly used” as encompassing cost, not just volume (eg, rheumatologists excluded from consideration practices that were not very costly to the health care system).
-
Use of the Delphi method4 as the overarching methodology for identifying a top-five list.
The three specialties reviewed the US and Canadian Choosing Wisely lists as part of their development process, but this was not a substitute for formulating their own lists, as not every international practice is relevant to Australia. For example, performing whole-body bone scans (eg, scintigraphy) for diagnostic screening for peripheral and axial arthritis is included in the Canadian rheumatology list but is not material to Australia.
Three Delphi method case studies
EVOLVE recommends use of the Delphi method for identifying low-value care practices, in keeping with initiatives elsewhere.5 This survey-based approach derives consensus based on purposive sampling of experts in the field of interest, panellist anonymity and iterative questionnaire presentation.4
There were three subtle differences in the way the method was applied by the specialties:
-
Both geriatric medicine and palliative medicine working groups consulted their memberships early in the process to seek comment on provisionally identified practices and suggest new ones. Only after processing membership feedback and refinement of the provisional list was an evidence review conducted.
-
Both geriatric medicine and palliative medicine working groups shortlisted their identified practices by requiring respondents to assign scores to each practice based on multiple criteria. Geriatric medicine used seven criteria, while palliative medicine used three. “Strength of evidence”, “significance” and “opportunity to make a difference” were criteria common to both.
-
Rheumatology recruited additional members (including three trainees) into the working group so they could invest effort in building on the RACP’s initial evidence review. With this larger working group, they could break into smaller teams and assign to each team a practice for further research. The evidence was summarised in an online survey distributed to the broader membership, with links to a full discussion of the evidence embedded in the survey questions.
Remaining challenges
Notwithstanding EVOLVE’s established principles and methods, some challenges remain.
First, without a requirement for compulsory participation, there is an element of self-selection in participation in specialty working groups and surveys. It is unclear whether this will lead to bias in the list of practices compiled for investigation and final shortlisting.
Second, there may be a risk of limited buy-in by specialty members if survey participation rates are low or if consensus cannot be reached, potentially reducing the impact of EVOLVE lists on clinician behaviour. The rheumatology working group aims to overcome this by encouraging high survey participation and by requiring that each top-five list practice be selected by at least 70% of survey respondents, in the hope that this represents a sufficiently high threshold for buy-in.
Third, ensuring that practices being considered are commonly used or increasing in use is difficult. For some practices, regularly collected publicly available data are incomplete (eg, MBS data that do not cover all hospital-provided services or do not provide sufficiently detailed breakdowns by indication). In other cases, the judgement of survey respondents or working group members (most of whom are in active clinical practice) was relied upon to remove practices considered irrelevant because of low levels of use.
Fourth, due to the clinical expertise required to formulate EVOLVE lists, the process is specialist-dominated. Nevertheless, achieving buy-in from consumers and non-RACP clinicians is critical, as sustaining changes in clinical practice requires cooperation from these groups. This task will benefit from support from Choosing Wisely Australia, led by NPS MedicineWise, to which EVOLVE is a contributor.
Finally, implementation of the EVOLVE recommendations into practice will be the greatest challenge. A recent study of the Choosing Wisely campaign in the US found significant declines being achieved in only two of seven low-value services identified by the campaign.6 Translating the EVOLVE recommendations into clinical practice requires both consumer and clinician education and a systemic cultural shift towards high-value care. This might be achievable if there is a systematic and coordinated approach, but a substantial investment in time and support may be required to ensure that the aims of EVOLVE are achieved and are sustainable over time.
Insights relevant to the MBS Review Taskforce
The MBS Review Taskforce’s early work has focused on identifying “obsolete” MBS items.7 EVOLVE’s focus is on reducing low-value care. Use of the 23 obsolete items identified by the Taskforce will, by definition, be declining, so aiming to reduce their use will have minimal impact. By contrast, one of the EVOLVE criteria is that the practice examined is commonly used or growing in usage.
A critical EVOLVE insight is that few practices are unambiguously low value for all clinical indications, and low-value care is contextual. Hence, there will be few genuinely low-value clinical practices that could be reduced by deleting particular MBS items.
Clinical practice is more likely to be improved by ensuring tests and treatments are targeted at people with appropriate clinical indications. The following low-value practices identified by the three specialties illustrate the importance of this:
-
use of ultrasound imaging to guide glucocorticoid injections into the shoulder or lateral hip compared with non-image-guided injections (rheumatology)
-
use of antipsychotics as the first choice to treat behavioural and psychological symptoms of dementia (geriatric medicine)
-
use of oxygen therapy to treat non-hypoxic dyspnoea in the absence of anxiety and as routine treatment at the end of life (palliative medicine).
Although these three practices identify low-value applications of ultrasound imaging, antipsychotics and oxygen therapy for specific indications or groups, this does not justify withdrawing subsidies entirely from these tests and treatments, as they are valuable in other clinical contexts.
While some clinical change can be induced by restricting conditions under which particular MBS items are covered, our examples also illustrate the limits of this approach. First, there are tests and treatments that are at risk of being misused but are not funded by the MBS. Second, imposing additional restrictions on the use of MBS items does not guarantee adherence unless proof of correct indication is required.
Financial incentives have been found to have limited effectiveness in driving sustained changes in clinical practice.8 Thus, it is likely that a systems-based approach employing multiple complementary strategies is needed. Initiatives like EVOLVE, that create endorsed and recognised peer judgements on what is good clinical practice, combined with other strategies such as the current MBS Review and mechanisms to improve clinician and consumer understanding of what constitutes low-value care, are needed. Working together, such strategies may shift clinician behaviour and consumer preference towards opting for the most appropriate evidence-based tests and treatments.
Statins and tendinopathy: a systematic review
The prescription of statin medications has increased over the past two decades.1 In 2006, 157 million prescriptions for statins were issued in the United States, grossing $16 billion.2 Since clinical release of the first statin, lovastatin, in 1987, considerable time has elapsed for post-marketing surveillance. Overall, statins are considered to have a well documented safety profile, with less than 2% of patients treated with atorvastatin ceasing the medication because of a drug-attributable adverse event.3 One area of interest in post-marketing surveillance of statins has been the musculoskeletal system,4,5 with conjecture that statins may contribute to tendinopathy.
Two significant attempts have been made to relate tendinopathy to statin therapy.6,7 In a retrospective observational case series of 31 French pharmacovigilance centres to determine the rates of tendon rupture or tendonitis among statin users, Marie and colleagues7 observed an increase in the number of reported tendinous complications, with eight reports from 1990 to 1995, and 56 from 2001 to 2005. However, this may simply reflect higher prescribing rates of statin therapy over time. Hoffman and colleagues reported pooled data for tendon and joint adverse events among patients using statins,6 but pooling of different structural outcomes makes interpretation problematic. Neither study included a comparator group to determine the rate of tendinopathy occurring among people not using statin therapy.6,7 Therefore, no conclusion can be made regarding whether the rates of tendinopathy while using statin therapy differed from the background rate of tendinopathy in the general community.
To assess this question, we performed a systematic review and examined the evidence for potential causation using the Bradford Hill criteria.
Methods
This systematic review was conducted according to the 2009 PRISMA statement.8
Search strategy and methodological quality
We searched four databases (Ovid MEDLINE, CINAHL Plus, PubMed and Embase) for the period January 1966 to October 2015 using the search terms described in Appendix 1. The search was limited to adult human studies published in the English language. We also searched the reference lists of included studies. To be included in this systematic review, studies had to report a comparison of a tendinopathy outcome between participants exposed to statins and those not exposed to statins. Study designs eligible for inclusion were randomised controlled trials and cross-sectional, cohort or case–control studies (Appendix 2). We excluded case reports and case series from the best evidence synthesis but include these reports in our analysis of the Bradford Hill criteria for causation.
Two independent reviewers (AJT and SREB) screened the identified articles and scored the methodological quality of included articles using the adapted scoring system of Lievense and colleagues9,10 (Appendix 3). Each methodological quality item was scored as positive (1), negative (0) or unclear (?), with a maximum possible score of 100%. Where the reviewers disagreed and could not achieve a consensus, a third reviewer (FMC) gave a final judgement. “High quality” was defined as achieving a score above the mean of all quality scores. We also used the Risk of Bias Assessment Tool for Nonrandomized Studies (RoBANS) to assess methodological quality of the included studies.11
Full-text articles that were excluded are listed in Appendix 4.
Best evidence synthesis
We used best evidence synthesis to summarise the data (Box 1).10,11 Studies were ranked according to their design, with cohort studies considered to be a higher level of evidence than case–control and cross-sectional studies. The level of evidence of studies was determined in conjunction with the quality score calculated for each study. For instance, “limited evidence” was concluded if generally consistent findings were demonstrated in a single cohort study, one or two case–control studies or multiple cross-sectional studies. In contrast, “strong evidence” was supported by generally consistent findings in multiple high-quality cohort studies.
Bradford Hill Criteria for causation
We used the Bradford Hill criteria to determine whether there was adequate evidence of a causal relationship between statin use (the risk factor) and tendinopathy (the disease).12
Results
We identified four studies for inclusion in this systematic review (Appendix 2).13–16 These studies were published between 2009 and 2015 (Box 2). Three were cohort studies and one was a case–control study. One study was conducted in Taiwan13 and all others in the US. In two studies, patients were recruited as cases from hospital databases,14,15 with the other studies collecting data from insurance databases.13,16 When people who were not exposed to statins were recruited, they were age- and sex-matched from the same institute as the index case.14,16 Tendon rupture was the primary outcome in three studies, and rotator cuff disease in the other.13
Definitions of statin exposure and tendinopathy
Definitions of statin exposure varied, including statin use in the 12 months preceding tendon rupture,14 or commencing therapy between 2003 and 2010 and having at least 1 year of continuous enrolment in a private insurance database.16 In the Taiwanese study, patients who used statins for at least 28 days were defined as users.13 In another study, medication histories were assessed at the time of surgical repair of the ruptured biceps tendon.15 No measure of compliance was recorded in any study.
In two studies, tendon ruptures were identified by code 727.xx in the International Classification of Diseases, 9th revision (ICD-9).14,16 In the other tendon rupture study, ruptures were identified historically, whereby spontaneous ruptures were defined as occurring during activities of daily living, and provoked ruptures during strenuous activities.15 Incident cases of rotator cuff disease were identified by ICD-9, clinical modification (CM) diagnosis codes 726.1, 727.61 or 840.4.13 The anatomical site of rupture varied, with one study focusing solely on the distal biceps tendon,15 while others pooled results from various anatomical sites.14,16
Characteristics of people with tendinopathy
In the largest study, Contractor and colleagues16 compared 34 749 people who commenced taking statins with 69 498 age- and sex-matched people who did not use statins. Beri and colleagues’14 case–control study compared 93 patients with tendon rupture with 279 age- and sex-matched controls without tendon rupture. Savvidou and colleagues15 reviewed a cohort of 104 patients with distal biceps tendon rupture. In the Taiwanese cohort, Lin and colleagues13 identified 25 621 patients with hyperlipidaemia, with 9.7% of these having rotator cuff disease. Participants’ ages ranged from 22 to 78 years. The proportion of women in the study populations ranged from 2% to 52.4%.
Is statin therapy associated with tendinopathy?
No study found an association between statin therapy and tendon rupture for the total population (Box 2).14–16 Beri et al14 demonstrated an association between tendon rupture and statin therapy for women only (odds ratio [OR], 3.76; 95% CI, 1.11–12.75; P = 0.043). However, only 22 people with tendon ruptures in their study had been exposed to a statin. Subgroup analyses based on sex further reduce the number of people with tendon ruptures exposed to statins and this result should therefore be treated with caution. The study authors repeated their work in an independent and larger cohort and found no predisposition by sex to statin-associated tendinopathy.16 In this larger study, subgroup analyses found that users of atorvastatin were at higher risk than those not taking statins (incidence rate ratio [IRR], 2.40; P = 0.0001), while simvastatin was protective (IRR, 0.77; P < 0.05). In the other tendon rupture study included in our review, although the authors concluded there was “a trend of association” between spontaneous distal biceps tendon ruptures and statin therapy (OR, 1.81; 95% CI, 0.56–5.84; P = 0.32), the direction of the result was changed when multivariate analyses adjusted for age (OR, 0.95; P = 0.94).15
In the study examining rotator cuff disease, exposure to simvastatin (hazard ratio [HR], 0.62; 95% CI, 0.54–0.71; P < 0.001), rosuvastatin (HR, 0.41; 95% CI, 0.35–0.49; P < 0.001) or other pooled statins (HR, 0.66; 95% CI, 0.60–0.72; P < 0.001) was associated with a reduced risk of rotator cuff disease.13
What does best evidence synthesis show regarding the association between statin therapy and tendinopathy?
The mean methodological score was 67%, with scores ranging from 42% to 83% (Appendix 5). Three of four studies were considered to be of high quality.13,14,16 The RoBANS found low risk of bias on most criteria in the studies (Box 3). Overall, the best evidence synthesis indicated limited evidence to conclude there was no association between statin therapy and tendon rupture. There was strong evidence that simvastatin was associated with a reduced risk of tendon rupture or rotator cuff disease.
What evidence is there for causation according to the Bradford Hill criteria?
We found that there was, at best, evidence for causation with respect to four of the Bradford Hill criteria: temporal relationship, strength of the association, reversibility and analogy (Box 4). There was poor evidence for the remaining criteria (plausibility, consistency and coherence) and no available evidence for specificity or a dose–response relationship. Overall, there was, at best, weak evidence for a cause–effect relationship. Nevertheless, the paucity of data in this field makes adequate interpretation of the Bradford Hill criteria difficult.
Discussion
In this review, we identified four studies examining the role of statin therapy in tendinopathy. Best evidence synthesis showed strong evidence that simvastatin reduced the risk of tendon rupture and rotator cuff disease, and limited evidence to conclude any adverse association between statin therapy and tendon rupture. Our findings suggest it is unlikely that statin therapy was a causative mechanism for tendon rupture among the total population of statin consumers.
The rate of tendon rupture among statin users was similar to the background rate in the general population.16 Contractor et al16 found that among just under 35 000 people who had commenced statin therapy, the rate of tendon rupture was small (n = 334) and no different from the rate (n = 800) observed in nearly 70 000 sex- and age-matched people not exposed to a statin. Indeed, in this large study, simvastatin users had a reduced risk of tendon rupture,16 a finding corroborated by another study demonstrating that statin exposure reduced the risk for rotator cuff disease.13
In 2003, 3.2 million people in France received statins, with only 13 statin-attributed tendon disorders reported in the national network of pharmacovigilance centres.7 As well as occurring infrequently, no study has reported an increased risk of tendinopathy related to statin therapy when analysing the total populations.14,16 While one study found an increased risk of tendon rupture in women,14 caution must be exercised when interpreting this subgroup analysis as the number of tendon ruptures for the total population was low.
It may be that case reports of tendon rupture among statin users gained notoriety because of the extreme nature of ruptures (eg, bilateral tendon ruptures) (Box 5).21–25 However, many of these reports are complicated by other risk factors for tendinopathy, such as hypercholesterolaemia,26,27 diabetes mellitus,28,29 fluoroquinolone use20,24 or extreme exertion.21 Higher serum cholesterol concentrations are associated with a thicker Achilles tendon,26,27,30,31 with localised oedema and inflammation, interfering with function and possibly contributing to tendon rupture.32,33 It has been found that after treatment with atorvastatin for 12 months, Achilles tendon thickness regressed among 15 heterozygous patients with familial hyperlipidaemia and tendon xanthoma.34 It is therefore difficult to distinguish whether tendinopathy was a result of drug intervention or whether it was the pathological process necessitating statin therapy that increased the risk of tendinopathy (ie, confounding by indication). Indeed, as part of the metabolic syndrome, people with dyslipidaemia often have co-existing hyperuricaemia and diabetes mellitus, factors recognised to independently increase the risk of tendinopathy.13,28,35,36 An Australian study found that, compared with 5159 age- and sex-matched controls without diabetes, there was a greater risk of tendon rupture requiring hospitalisation among people with type 2 diabetes (n = 1296).36 In the limited number of studies we found examining tendinopathy and statin use, three made a concerted effort to adjust for confounders such as diabetes mellitus or hyperuricaemia.13,14,16 Likewise, two studies found that no patients who had ruptured tendons had been exposed to fluoroquinolones.14,15 While Contractor et al16 did not report the number of people exposed to fluoroquinolones, this was a factor in the comorbidity index used in their regression analyses.
Another potential limitation to better understanding the possible association between statin therapy and tendon-related disorders is the heterogeneity among studies. For instance, Beri et al14 pooled tendon ruptures from multiple anatomical sites. It may be that the risk of tendon rupture is site-specific. Savvidou et al15 circumvented this limitation by only examining distal biceps tendon rupture, concluding that there was a trend towards increased spontaneous distal biceps tendon ruptures in patients taking statins. However, this trend was not statistically significant, and results changed direction when age was included in the multivariate analyses.
Our systematic review has several limitations. We identified only a small number of studies, and no randomised controlled trials. Studies varied considerably in their methodology, with three studies examining tendon rupture as the primary outcome and only one examining rotator cuff disease. Moreover, different clinical populations (eg, patients in university hospitals, private insurance databases and microsurgery institutes) exposed to various statins, with different follow-up times, as well as variations in handling of potential confounders and tendon rupture outcomes at different anatomical sites, contributed to the heterogeneity of studies. Along with the absence of randomised controlled trials, this precluded a meta-analysis of these data. We have instead used a best evidence synthesis approach,9,10 in which studies above the mean quality score of all available evidence are deemed high quality. This is a limitation, as even among a collection of generally poor studies, half will be deemed high quality. Although this may influence best evidence synthesis, it is a true reflection of the available published literature and this is the first systematic review of this topic. We also limited our search strategy to studies written in English. However, this is unlikely to have biased our results, as we identified no abstracts in other languages. Moreover, we also used the RoBANS, which found low risk of bias on most criteria.
Conclusions
On the basis of the limited data available, tendon rupture while using statin therapy is an infrequent occurrence, and one high-quality study demonstrated that event rates were no different to the background rate in the general population. Indeed, two high-quality cohort studies provided strong evidence that simvastatin reduces the risk of tendinopathy. There is limited evidence for causality between statin therapy and tendon rupture.
Box 1 –
Criteria for determining the level of evidence in best evidence synthesis*
|
Level of evidence |
Criteria for inclusion in best evidence synthesis |
||||||||||||||
|
|
|||||||||||||||
|
Strong evidence |
Generally consistent findings in:
|
||||||||||||||
|
Moderate evidence |
Generally consistent findings in:
|
||||||||||||||
|
Limited evidence |
Generally consistent findings in:
|
||||||||||||||
|
Conflicting evidence |
Inconsistent findings in < 75% of studies |
||||||||||||||
|
No evidence |
No studies could be found |
||||||||||||||
|
|
|||||||||||||||
Box 2 –
Epidemiological evidence examining statin use and tendinopathy
|
Study |
Design and setting |
Population |
Results |
Conclusion |
Quality score |
||||||||||
|
|
|||||||||||||||
|
Beri et al (2009)14 |
Case–control study at Michigan State University (2002–2007) |
93 cases (29 women) and 279 sex- and age-matched controlsExposure defined as statin use in the 12 months preceding tendon rupture |
No difference between cases and controls in the rate of statin use (OR, 1.10; 95% CI, 0.57–2.13) after adjusting for diabetes, renal disease, rheumatological disease and steroid useStatin exposure was a significant risk factor for tendon rupture in women (OR, 3.76, 95% CI, 1.11–12.75) but not in men (OR, 0.66; 95% CI, 0.29–1.51) |
No association between statin use and tendon rupture for total populationSubgroup analysis suggests that women with tendon rupture were more likely to be taking statins |
69% |
||||||||||
|
Savvidou et al (2012)15 |
Retrospective observational study at Institute for Hand and Microsurgery, Louisville, Kentucky (2004–2010) |
104 patients (98% male; mean age, 47 years; age range, 22–78 years) treated for distal biceps tendon rupture between 2004 and 2010Spontaneous rupture (n = 19) defined as occurring during activities of daily livingProvoked rupture (n = 85) defined as occurring during strenuous activities |
Of the 104 patients, 19 were taking statins (5 in spontaneous rupture and 14 in provoked rupture group)No significant association between odds for spontaneous distal biceps tendon rupture and taking statin (OR, 1.81; 95% CI, 0.56–5.84; P = 0.32). When adjusted for age: OR, 0.95; P = 0.94 |
Authors concluded a trend of association between spontaneous distal biceps tendon rupture and statin therapy |
42% |
||||||||||
|
Contractor et al (2015)16 |
Retrospective cohort study using private insurance database |
34 749 people commencing statins after the beginning of the study period, and 69 498 age- and sex-matched adults not exposed to statins (47.7% female; mean age, 47.6 years, age range, 30–64 years) |
Rate of tendon rupture
No difference in tendon rupture between users and non-users of statins after adjusting for comorbidity index, age and sex: IRR, 1.13 (95% CI, 0.98–1.29)The most commonly prescribed drug was simvastatin (57.4%; 19 902) followed by atorvastatin (19.0%; 6596), lovastatin (16.5%; 5736) and pravastatin (5.2%; 1794), with others accounting for less than 2%. When assessing the risk with individual statins, atorvastatin users had a significant risk of tendon rupture compared with controls (IRR, 2.40; P = 0.0001). Conversely, simvastatin use was associated with a lower risk of rupture (IRR, 0.77; P < 0.05) |
No association between statin use and tendon rupture when all statins examined |
75% |
||||||||||
|
Lin et al (2015)13 |
Prospective Taiwanese cohort study of patients diagnosed with hyperlipidaemia in 2000 and followed for 11 years (with or without statin use), from the National Health Insurance Research Database |
25 621 patients diagnosed with hyperlipidaemia (52.4% female; mean ± SD age, 57.6 ± 12 years; age range, 30 to ≥ 70 years) |
Rotator cuff disease present in 2475 patients (9.7%) with hyperlipidaemia. In patients with hyperlipidaemia, statin use was associated with a lower risk of developing rotator cuff disease when compared with no statin use:
|
Statin use might provide protection against rotator cuff disease in patients with hyperlipidaemia, independent of age, sex and diabetes status |
83% |
||||||||||
|
|
|||||||||||||||
|
OR = odds ratio. IRR = incidence rate ratio. HR = hazard ratio. |
|||||||||||||||
Box 3 –
Risk of Bias Assessment Tool for Nonrandomized Studies11
|
Bias type |
Description |
Risk of bias |
|||||||||||||
|
Beri et al (2009)14 |
Savvidou et al (2012)15 |
Contractor et al (2015)16 |
Lin et al (2015)13 |
||||||||||||
|
|
|||||||||||||||
|
Selection of participants |
Selection bias caused by inadequate selection of participants |
Low |
Low |
Low |
Low |
||||||||||
|
Confounding variables |
Selection bias caused by inadequate confirmation and consideration of confounding variable |
Low |
High |
Low |
Low |
||||||||||
|
Measurement of exposure |
Performance bias caused by inadequate measurement of exposure |
Low |
Low |
Low |
Low |
||||||||||
|
Blinding of outcome assessment |
Performance bias caused by inadequate blinding of outcome assessment |
Unclear |
Unclear |
Unclear |
Unclear |
||||||||||
|
Incomplete outcome data |
Attrition bias caused by inadequate handling of incomplete outcome data |
Low |
Unclear |
Low |
Low |
||||||||||
|
Selective outcome reporting |
Reporting bias caused by selective reporting of outcomes |
Low |
Low |
Low |
Low |
||||||||||
|
|
|||||||||||||||
|
|
|||||||||||||||
Box 4 –
Evidence for a causal relationship between statin therapy and tendinopathy, according to the Bradford Hill criteria
|
Bradford Hill criterion and description |
Evidence supporting or refuting a causal relationship between statin use and tendinopathy |
||||||||||||||
|
|
|||||||||||||||
|
Temporal relationshipThis is an essential criterion. For a possible risk factor to be the cause of a disease, it must come before the disease. This is generally easier to establish from cohort studies than from cross-sectional or case–control studies, when measurements of the possible cause and the effect are made at the same time |
Contractor et al (2015)16
Lin et al (2015)13
Therefore, there is no supportive evidence to fulfil a temporal relationship, other than in one subgroup analysis for atorvastatin use. Indeed, temporal relationship suggests a reduced risk of tendon rupture and rotator cuff disease in simvastatin users |
||||||||||||||
|
PlausibilityA risk factor associated with a disease is more likely to be the cause of the disease if the association found is consistent with knowledge obtained from other sources, such as animal experiments and experiments on biological mechanisms. However, this criterion must be used with care as a lack of plausibility may simply reflect a lack of scientific knowledge |
de Oliveira et al (2013)17
Ferreira et al (2014)18
|
||||||||||||||
|
ConsistencyIf similar results have been found in different populations using different study designs, the association is more likely to be causal as it is unlikely that all studies were subject to the same types of errors (chance, bias or confounding). However, a lack of consistency does not exclude a causal association, as different exposure levels and other conditions may reduce the impact of the causal factor in certain studies |
Contractor et al (2015)16
Marie et al (2008)7 |
||||||||||||||
|
Strength of an associationThe strength of an association is measured by the size of the relative risk. A strong association is more likely than a weak association to be causal, as a weak association could more easily be the result of confounding or bias |
Beri et al (2009)14
Contractor et al (2015)16
Lin et al (2015)13
|
||||||||||||||
|
Dose–response relationshipFurther evidence of a causal relationship is provided if increasing levels of exposure lead to an increasing risk of disease |
No available evidence |
||||||||||||||
|
SpecificityIf a particular exposure increases the risk of a certain disease but not the risk of other diseases, this is strong evidence in favour of a cause–effect relationship. However, one-to-one relationships between exposure and disease are rare, and lack of specificity should not be used to say that a relationship is causal |
No available evidence |
||||||||||||||
|
ReversibilityWhen the removal of a possible risk factor results in a reduced risk of disease, the likelihood that this association is causal is increased. Ideally, this should be assessed by conducting a randomised intervention trial. For many exposures or diseases, such randomised trials are not possible in practice |
Chazerain et al (2001)19
|
||||||||||||||
|
CoherenceThe suggested cause–effect relationship should essentially be consistent with the natural history and biology of the disease |
Tendinopathy can result from many mechanisms, including sport or heavy physical activity, hyperlipidaemia and medications such as fluoroquinolones |
||||||||||||||
|
AnalogyThe causal relationship will be further supported if there are similarities with other (well established) cause–effect relationships |
Other drug classes (fluoroquinolones) are a well established causal mechanism for tendinopathy20 |
||||||||||||||
|
|
|||||||||||||||
|
OR = odds ratio. IRR = incidence rate ratio. HR = hazard ratio. |
|||||||||||||||
Box 5 –
Case reports examining statin use and tendinopathy
|
Study |
Patient age, sex |
Comorbidities |
Statin |
Anatomical site |
Potential confounding |
||||||||||
|
|
|||||||||||||||
|
Carmont et al (2009)21 |
47 years, male |
Familial hypercholesterolaemia |
Simvastatin, 40 mg/dayExposure duration, 12 weeks |
Bilateral Achilles tendon rupture (simultaneous) |
Rock-climber who trained at least once per week for past 27 years, with familial hypercholesterolaemia |
||||||||||
|
Rubin et al (2011)22 |
58 years, male |
Hypertension |
Simvastatin, 80 mg/dayExposure duration, 4 years |
Complete bilateral quadriceps tendon rupture (simultaneous) |
None identified |
||||||||||
|
Celik et al (2012)23 |
56 years, male |
Abdominal aortic aneurysm repair |
Rosuvastatin, dose not statedExposure duration, 4 years |
Complete bilateral quadriceps tendon rupture while climbing stairs |
None identified |
||||||||||
|
Ganske et al (2012)24 |
58 years, male |
Recurrent otitis media treated with levofloxacin, 750 mg/day for 5 days (10 days before event) |
Simvastatin, 20 mg/dayExposure duration not stated |
Hip tendinopathy |
Exposure to fluoroquinolone |
||||||||||
|
Nesselroade et al (2010)25 |
56 years, male |
Nil |
Atorvastatin, 20 mg/dayExposure duration, 3 years |
Bilateral quadriceps tendon rupture (simultaneous) |
Football referee |
||||||||||
|
|
|||||||||||||||
|
|
|||||||||||||||
A case of bilateral endogenous bacterial endophthalmitis from Streptococcus pneumoniae bacteraemia
Clinical record
A 55-year-old woman was admitted to an orthopaedic unit of a metropolitan hospital in Australia with right shoulder septic arthritis. She had been experiencing 3 days of right shoulder pain, fevers, rigors and delirium. These occurred in the context of a right rotator cuff repair 3 months previously. Her medical history included hypertension, hypercholesterolemia and an L4-S1 spinal fusion for lumbar spine degeneration. Her medications included hydrochlorothiazide, olmesartan and atorvastatin.
On examination, she was febrile with a temperature of 38.9°C, with a painful, erythematous and swollen right shoulder with movement limited by pain. Her chest and abdominal examinations were normal. Her relevant admission blood test results were white cell count, 30 × 109/L (reference interval [RI], 4–11 × 109); neutrophil count, 21.9 × 109 (RI, 2–8 × 109); and C-reactive protein level, 438 mg/L (RI, 0–5 mg/L). Platelet, haemoglobin, uric acid and blood sugar levels were all normal. Her shoulder and chest x-rays were unremarkable. Several sets of blood cultures as well as a shoulder aspirate showed gram-positive cocci and she was empirically commenced on intravenous (IV) flucloxacillin 2 g four times a day and vancomycin 1.5 g twice daily.
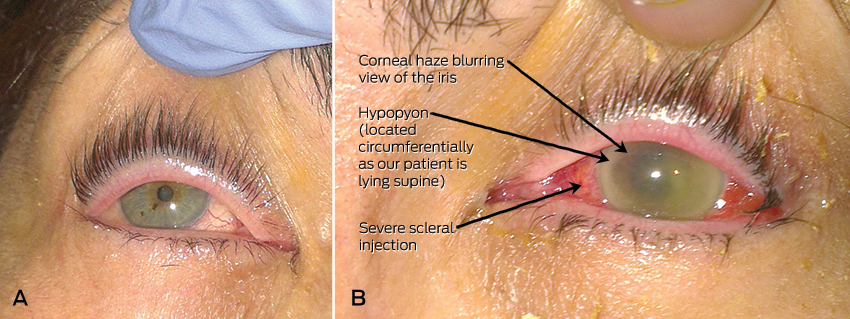
Shortly after admission, she revealed that she had been experiencing 3 days of blurred vision and pain in her left eye. Further history revealed that aside from mild myopia, her vision was previously normal and she had never undergone any ophthalmic procedures. Her right eye was asymptomatic and appeared slightly injected but otherwise normal on initial assessment (Figure, A). However, her left sclera was severely injected, and the pupil was fixed and mildly dilated with corneal and anterior chamber haze and a circumferential hypopyon (Figure, B). Visual acuity (VA) was 6/12 in her right eye, but only hand movements were appreciated in her left eye. Fundoscopy was not possible on the left eye owing to the severity of the corneal haze; however, in the right eye, fundoscopy showed a vitreous haze.
An urgent assessment by an ophthalmologist found that she had bilateral endogenous bacterial endophthalmitis. Despite appearing mostly unremarkable on external inspection, the right eye was deemed to be in the early stages of infection owing to the presence of vitreous haze on fundoscopy. After aqueous humour samples were taken, she was given bilateral intravitreal antibiotic injections of 1 mg vancomycin and 2 mg ceftazidime. This was followed up with intravitreal injections of 1 mg vancomycin every second day for three further doses. The cultures from her shoulder, blood and eyes all grew Streptococcus pneumoniae. Her antibiotics were changed to IV benzylpenicillin and vancomycin; IV azithromycin was also added, as evidence has shown that it has a survival benefit in S. pneumoniae infections.1 Further investigations performed to exclude immunosuppression and locate the source of the septicaemia included HIV, tuberculosis and hepatitis serology; glycated haemoglobin testing; vasculitic screening; a transthoracic echocardiogram; computed tomography of the chest, abdomen and pelvis to look for malignancy or abscesses; and a magnetic resonance imaging scan of her lumbosacral spine to exclude infection of the implants from her spinal fusion. The results of these tests were unremarkable and no cause was found to explain the S. pneumoniae septicaemia.
Twelve days after admission, she was transferred to another centre with a vitreoretinal unit, where she underwent a left vitrectomy to debride the posterior chamber of her eye. Unfortunately, the vision in her left eye only made a small recovery and, 6 months later, the VA was limited to counting fingers. Fortunately, VA in her right eye remained at its baseline and she has been managing well in the community.
Bacterial endophthalmitis is an infection of the eye involving the aqueous and/or vitreous humour. It is an ophthalmic emergency and requires urgent treatment to prevent permanent blindness. The vast majority of cases are due to exogenous inoculation of microbes into the eye via trauma, surgery, intravitreal injections or an invasive corneal infection. Rarely, in 2–6% of cases, it can occur from haematogenous spread of organisms to the eye, which is referred to as endogenous or metastatic endophthalmitis. Endogenous spread is associated with immunosuppressive predisposing factors such as diabetes or malignancy in 90% of cases.2
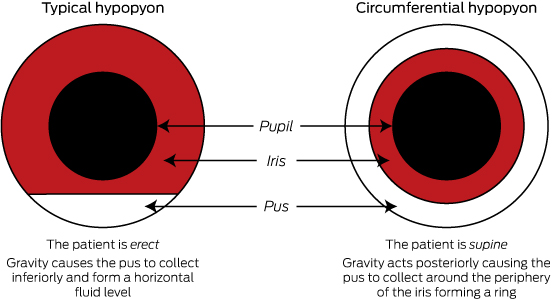
Patients usually experience 12–24 hours of eye pain and decreasing vision. They are rarely systemically unwell. However, patients with the endogenous variant are usually unwell from the underlying bacteraemia. On examination, there is often conjunctival injection or oedema of the cornea. VA will be decreased. Slit-lamp examination of the anterior chamber may show cells and flare (small floating particles and a generalised haziness, respectively, in the anterior chamber representing inflammation). As in this patient, it may be also be associated with a hypopyon. Hypopyon is pus in the anterior chamber of the eye which will collect inferiorly and form a horizontal layer in an erect patient (Figure, B shows a circumferential hypopyon in our patient as she was supine at the time she was photographed; the Box highlights the difference between hypopyon in erect and supine patients). On fundoscopy, there is usually poor visualisation of the retina secondary to vitreous haze.2,3 The diagnosis can be confirmed via aqueous or vitreous culture, but it should be noted that history and examination are the main diagnostic tools, as a negative culture cannot reliably exclude the condition.4
Bacterial endophthalmitis is usually exogenous and postoperative. It is most commonly caused by coagulase-negative staphylococci (the most common normal flora of the ocular surface).5 The organisms for the endogenous form depend on the underlying infection. A major study showed only 30% of patients with streptococcal endophthalmitis had a final VA of 6/30 or better, which is worse than for Staphylococcus aureus, gram-negative bacteria (50% for both) and coagulase-negative staphylococci (80%).6 Regardless of the organism involved, the strongest indicator of visual prognosis is VA at presentation.5
Treatment of endophthalmitis requires prompt referral to an ophthalmology team. Treatment of bacterial endophthalmitis involves intravitreal antibiotic injections as the intravenous route does not deliver sufficient concentrations to the relatively avascular posterior chamber of the eye. Empirical therapy is usually vancomycin with ceftazidime or amikacin. This is often combined with vitrectomy, which debrides the vitreous, reducing the bacterial load. Patients should also be treated with systemic antibiotics for sufficient duration to clear the underlying infection.7
Bilateral endogenous bacterial endophthalmitis is very rare, with one study estimating that only around 12% of patients with endophthalmitis had both eyes infected.2 A rare bilateral case like ours, where one eye is severely infected and the other is in the early stages of infection, serves to demonstrate the importance of early diagnosis, as once vision has been lost it is far less likely to return. Further, our case is unusual in that no immunosuppression was found in a relatively young patient.
Lessons from practice
-
Endogenous bacterial endophthalmitis can rapidly cause blindness and should be considered in all bacteraemic patients with decreased visual acuity or ocular pain.
-
Risk factors include diabetes, malignancy and immunosuppression.
-
Early diagnosis is important as visual acuity on diagnosis is the strongest indicator of final visual prognosis.
-
Early ophthalmology involvement and intravitreal antibiotic injections are essential, as intravenous therapy alone will not deliver adequate concentrations to the relatively avascular vitreous.
Vertebroplasty is not a do-not-do treatment
Vertebroplasty has been controversial but remains clinically useful and new evidence awaits publication
Duckett and colleagues have classified vertebroplasty as a do-not-do treatment.1 They referenced two randomised controlled trials (RCTs)2,3 as definitive proof of this. However, the authors failed to heed our clinical opinion published in the MJA that these two trials were “not relevant to the patient group that we treat with vertebroplasty”.4 We have the largest clinical vertebroplasty experience in Australia, yet our published advice was apparently ignored. In the article by Duckett and colleagues, Box 1 illustrated the selection process that the authors used to determine do-not-do procedures. The process supposedly excluded evidence which was “contested” or “which was not supported by consulted clinical experts”. Accordingly, vertebroplasty should have been deleted from the list.
The authors used the United Kingdom National Institute for Health and Care Excellence (NICE) for clinical guidance. Current NICE guidance5 states that “vertebroplasty and kyphoplasty can be considered appropriate interventions for people with recent, unhealed osteoporotic vertebral compression fractures in whom the pain is severe and ongoing despite optimal pain management”.
From 1208 potential treatments, the authors excluded 1200, leaving five apparently incontrovertible do-not-do treatments. The fact that at least one of the five is wrongly included (by the authors’ own criteria) demonstrates the failure of the proposed model and the danger of adopting this kind of formula to influence clinical practice in hospitals.
The evidence for and against vertebroplasty is inconclusive. There is disparity in measured outcomes between blinded RCTs2,3 of vertebroplasty for fractures up to 12 months old and a larger, open-label RCT6 of fractures less than 6 weeks in duration. The blinded trials found no significant benefit of vertebroplasty over placebo, whereas the open-label RCT found significant benefit of vertebroplasty over conservative care. This disparity is well described in the NICE guidance.5
For the past 10 years, my vertebroplasty practice has been confined to treating fractures less than 6 weeks old.7 It is clear to me that the published blinded trials tested a different approach and are not relevant to the patient group that my practice treats with vertebroplasty for two principal reasons: the fractures were mostly non-acute; and the volume of cement used in these trials (2.6 cm3 on average in both trials) would have been insufficient to stabilise an acutely collapsing vertebral fracture.
Attempting to answer the acute fracture conundrum, the authors of the blinded RCTs published a meta-analysis of 52 patients from both trials with fractures less than 6 weeks duration.8 Only outcomes at 2 weeks and 1 month were presented and the evidence is hardly definitive.
The onus was placed on vertebroplasty practitioners to provide high-quality blinded data in this group of patients. For this purpose, my co-investigators and I embarked on the Vertebroplasty for Acute Painful Osteoporotic fractURes (VAPOUR) trial.9 Five years ago, we reconfigured the protocol from the INvestigational Vertebroplasty Efficacy and Safety Trial (INVEST),2,10 the larger of the two blinded vertebroplasty trials. We excluded crossover, which was permitted at 1 month in INVEST. We changed the selection criteria to include only fractures less than 6 weeks duration (average fracture age in the VAPOUR trial was 2.6 weeks compared with 18 weeks in INVEST) with pain scores greater than 7/10 and with either magnetic resonance imaging or single-photon emission computed tomography evidence of acute fracture. In-patients, already hospitalised with acute fractures, comprised 59% of the VAPOUR trial enrolment but were excluded in INVEST. The procedural technique was different in the VAPOUR trial, where we attempted maximum fill of the vertebral body to stabilise the fracture and prevent ongoing collapse. The average cement volume of 7.5 cm3 in the VAPOUR trial was three times that in INVEST. The method of blinding and data collection was similar for the two trials.
Our trial team included four Sydney centres with established vertebroplasty programs. The VAPOUR trial completed enrolment of 120 patients in December 2014 and is the largest RCT and the only acute fracture RCT of vertebroplasty in Australia. Statistical assessments of outcomes are nearing completion and the results of the trial will soon be published.
Ten years of publicly funded biological disease-modifying antirheumatic drugs in Australia
Access to high-cost medicines through the Australian Government’s Pharmaceutical Benefits Scheme (PBS) poses a number of challenges for the National Medicines Policy, which aims to provide timely access to medicines at a cost individuals and the community can afford. Biological disease-modifying antirheumatic drugs (bDMARDs) for rheumatoid arthritis (RA) were among the first highly accessed, high-cost drugs to be subsidised in Australia. To restrict their use, a unique subsidy scheme was developed by the Pharmaceutical Benefits Advisory Committee (PBAC), whereby physicians complete written authorities documenting both eligibility to initiate and response to permit continuation.1 Similar models have been adopted for other high-cost medications (eg, trastuzumab),2 but there is continuing debate on whether the medicines are available for all who need them, and what constitutes a “worthwhile” response for the individual and for the community who pay.3,4 In this article, we reflect on bDMARD use and the expenditure for newer bDMARDs for RA (abatacept, tocilizumab, certolizumab pegol and golimumab) (our methods are described in the Appendix). We suggest that an electronic database of data submitted for subsidised bDMARD use could promote the quality use of medicines (QUM) and improve cost expenditure predictions for high-cost drugs.
bDMARDs on the PBS for rheumatoid arthritis
Etanercept and infliximab were PBS listed for RA treatment in 2003, and eight bDMARDs are now available (Box 1). These agents cost between $15 000 and $25 000 per patient per annum, and use has increased such that expenditure was about $383 million in 2014 (Box 2).
Etanercept and adalimumab remain the most commonly prescribed bDMARDs, collectively accounting for 64% of bDMARD use in 2013–2014 (Box 3). The cost of the available bDMARDs on the PBS for RA treatment is relatively uniform, with each bDMARD having been added on a cost minimisation basis compared with a competitor.5 The dispensing cost for each bDMARD (for the most dispensed item code), and the PBS-reported dispensing price for the maximum quantity, are shown in Box 4.6
The subsidy process
In 2003, consultation between stakeholders (the PBAC, pharmaceutical companies, rheumatologists and consumers) resulted in access criteria (to limit access to those for whom less expensive options had been inadequate to achieve acceptable disease activity) and response criteria (based on ACR50 responses [at least a 50% improvement in the American College of Rheumatology score], to ensure only those responding received continued treatment).1 Limitations to access were also thought warranted given concerns regarding the possibility that bDMARDs might increase the risk of lymphoma. After review, these restrictions were relaxed in 2010 (Box 5).7 The cost of etanercept and adalimumab has decreased from about $1880 per patient per month in 2003 to $1760 per patient per month in 2014, reflecting a price reduction to achieve a more acceptable cost–effectiveness ratio when the initiation criteria were modified.7
After failure of five bDMARDs, no further access to biological agents within a lifetime is allowed,7 so patients may access two tumour necrosis factor (TNF) blockers and each of the bDMARDs with distinct alternative actions. The proportion of patients who exhaust all bDMARD options is likely to be small.
Formal evidence that strict initiation and continuation criteria contain gross drug acquisition costs and improve the overall cost–benefit ratio has face validity, but formal proof is lacking and difficult to establish. The need to interpret complex rules and to complete onerous paperwork for access, incidentally or by design, is a disincentive to the promiscuous use of expensive agents. Nonetheless, assessing disease activity at baseline and after defined periods introduces to clinical practice formal assessment of RA disease activity, hitherto reserved for clinical trials. It is thus difficult to argue that such procedures should not be used in routine clinical practice, if for no other reason than to avoid resource expenditure on patients failing to fulfil response criteria and who may benefit from alternative treatment.
Circumventing restrictions
As high disease activity at baseline and subsequent demonstrable clinical response is required for initial and continued bDMARD therapy, concerns are raised regarding those not meeting initiation criteria, or not meeting response thresholds despite improvement.8 The ethical burden this places on rheumatologists is undefined; however, practices may be altered such that clinicians may be tempted to allow disease activity to increase before applying for bDMARD initiation or bias baseline joint counts.
Expenditure predictions and monitoring
To predict cost-effectiveness and the financial implications of listing a medication on the PBS, usage is estimated before listing,9 and the difference between estimated and actual usage is serially assessed after listing.10
In the 2 years following etanercept and infliximab listing (Box 1), the actual expenditure was $53 million (19% of predicted).11 This low uptake may have reflected concerns regarding safety, perceived efficacy, availability of the drugs and facilities to administer them, the complexity of the rules, and the administrative burden of applying for initiation and continuation of these agents. Since 2008, abatacept, tocilizumab, certolizumab pegol and golimumab have been introduced, and their use has steadily increased (Box 6). The estimated annual expenditure in the first 5 years for abatacept was $25 million (ie, 50 000 vials at $500 per vial) and less than $10 million for each of tocilizumab, certolizumab pegol and golimumab.12–15 Expenditure on abatacept was less than predicted for each of the first 5 years, but tocilizumab, certolizumab pegol and golimumab had a combined expenditure of about $93 million for the fifth year of their listing, 210% over the initial estimate. Over the same period, the combined annual expenditure on etanercept and adalimumab rose from about $110 to $227 million. The reasons for the differential uptake of bDMARDs are unclear. Given the lack of head-to-head studies to evaluate the relative efficacy of bDMARDs, ease of administration and access to appropriate facilities, growing experience with use and the (perceived or actual) benefit of alternative mechanisms of action may be important determinants of choice. Also, the respective marketing strategies of sponsoring pharmaceutical companies are likely to have had an effect.
Cost forecasting for bDMARDs has failed, highlighting the problem with estimations in their current format. It is unclear whether the cost underestimations for tocilizumab, certolizumab pegol and golimumab were due to a larger than expected switch rate, circumvention of restrictions or preference for newer bDMARDs rather than older agents. Recently, European League Against Rheumatism guidelines removed the recommendation that the first-line bDMARD should be a TNF inhibitor, instead stating that there was no preference.16 However, it does not appear that non-anti-TNF agents (bDMARDs with other mechanisms of action) were strongly preferred, since etanercept and adalimumab use continued to increase. It is also difficult to apportion the effect of changing PBS restrictions in 2010 on anticipated uptake.
The implications of inaccurate expenditure predictions on the National Medicines Policy can be significant. Initiation and continuation criteria are based on clinical trial data and inaccurate predictions could result from difficulty in translating trial evidence to practice. Underestimation of use could be due to a lower than expected commitment or response rates to existing conventional DMARDs and bDMARDs, and this has substantial long-term cost implications. Underestimation could reflect an unforeseen preference for the newer agents (compared with the older, similarly priced biological agents), which could lead to benign cost-shifting. However, the net effect is uncertain, since managed-entry agreements, which share the financial risks of underestimation between funders and sponsors of recently introduced high-cost drugs, are common. The details of such arrangements are not publicly available, and extend beyond the restriction criteria and cost predictions presented herein. For the 2013–14 financial year, the Australian government recovered $500 million from managed-entry agreements and, as such, the importance of inaccurate cost prediction, at least for the duration of these agreements, is uncertain.17 It is likely that price-capping agreements were in place for abatacept, tocilizumab, certolizumab pegol and golimumab throughout the investigated period, although details of recovered monies are not available.2
Biosimilars
Recently, biosimilars of etanercept and infliximab have offered promise for providing less expensive bDMARDs. Unlike small-molecule generic products that can lower price by as much as 90%, biosimilars have more complex development processes which result in post-translational modifications in structure. Typically, prices are 20%–30% lower than branded products.18 These factors can result in reluctance to switch from branded products that are “known” to work.18 However, clinicians and sponsors may be placing too strong an emphasis on post-translational differences, as branded agents can also exhibit functionally and/or immunologically important post-translational modifications over time.19 The price of a branded bDMARD undoubtedly includes a substantial profit margin to compensate for development costs. A biosimilar, however, can be developed in the knowledge that it is directed at an appropriate target. There must, therefore, be appropriate and proportionate incentives to develop both novel bDMARDs and cost-effective biosimilars. There is a need for regulatory agencies, funders and manufacturers of reference and biosimilar agents to strive to assemble clinical research data, and to achieve funding arrangements which increase access to the most effective biological products (be they branded or biosimilar) at a time when they will be most effective. Without this collaborative action, we may not see a substantial reduction in the cost of newer bDMARDs until patents expire on the small-molecule tyrosine kinase inhibitors that are not yet marketed for the treatment of RA in many countries.20
Future perspectives
A key feature of Australian programs for access to high-cost drugs is the need to provide clinical data before initiation and when evaluating response. These data could be used to examine real-world effectiveness and review expenditure predictions, with a view to developing better predictive models; however, these data have been inaccessible to date.1,21
Recent advents to allow access to a 10% sample of individualised PBS data are a step towards improved QUM monitoring. Yet, a real-world database of clinical and laboratory data could also have an important role. The information would, however, lack patient-reported data and quality-of-life outcomes. These shortcomings could be addressed in part by linkage with the Australian Rheumatology Association Database, which contains this information for a subset of the population.22 A real-world database would also present a potentially novel framework for nationwide research projects that could, for example, investigate the clinical utility of pharmacogenomic and therapeutic drug monitoring variables as a means to optimise bDMARD selection. It would have the potential to answer questions about bDMARD use that have remained unanswered over the past 10 years, some of which are listed in Box 7.
Like the novel bDMARDs introduced before them, bDMARD biosimilars will be among the first high-cost agents in their class to be publicly funded, and the developed funding models will lay the framework for accessing and pricing biosimilars in the future. Since their introduction is likely to result in only modest price reductions, relaxation of access criteria is unlikely unless acceptable reductions in cost–effectiveness ratios can be demonstrated. Funding of medicines currently relies on substantial cost reduction following the introduction of generics, which frees up funds that can be directed towards new medicines.
In this article, we have used bDMARDs to exemplify current and future challenges to achieving equitable access to high-cost drugs. A real-world database could also inform questions regarding effectiveness and appropriate access of other high-cost drugs that use similar written authority-based prescribing models. In turn, these data could inform future negotiations regarding what constitutes a fair price for the benefit accruing from the use of these agents.
Conclusion
The use of bDMARDs to treat RA, and the cost to taxpayers, has steadily increased in Australia since 2003. Despite the newer bDMARDs, etanercept and adalimumab remain the two most used agents. The impact of strict initiation and continuation rules on overall expenditure is uncertain, but is likely to have lessened as prescribers have become more familiar with procedures. Prediction of expenditure on newly introduced high-cost drugs remains inaccurate despite substantial experience with the systems for restricted access. Present and future opportunities that must be explored to ensure that the goals of the National Medicines Policy are met include the strategic development, regulation and use of biosimilars. These endeavours should include analyses of authority prescription data which have been collected over the past 10 years and continue to be collected with the objectives of refining access and improving cost–effectiveness ratios.
Box 1 –
Timeline of bDMARD addition and subsidisation for RA on the PBS*
bDMARD = biological disease-modifying antirheumatic drug. RA = rheumatoid arthritis. PBS = Pharmaceutical Benefits Scheme. * Anakinra is an interleukin 1 receptor antagonist that is classified as a “biological response modifier”.
Box 2 –
Use and service cost per year (combined cost to PBS and RPBS) for anti-TNF and non-anti-TNF bDMARDS for treating patients with RA in Australia, 2003–2014*
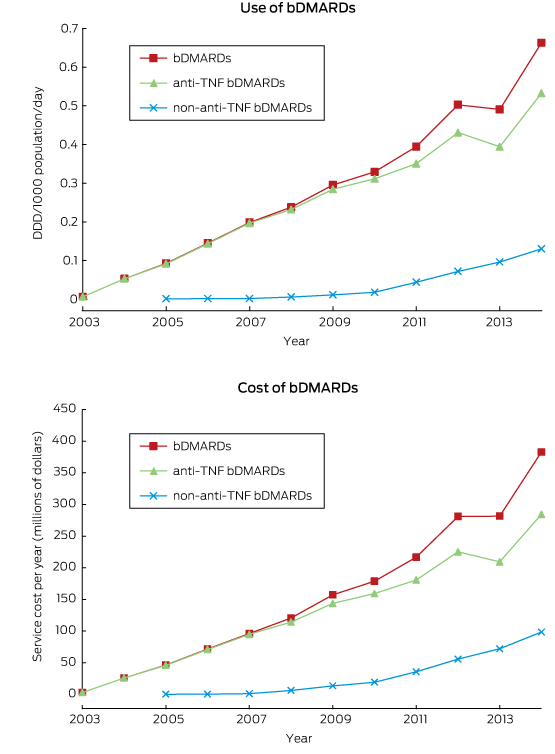
bDMARD = biological disease-modifying antirheumatic drug. PBS = Pharmaceutical Benefits Scheme. RPBS = Repatriation Pharmaceutical Benefits Scheme. TNF = tumour necrosis factor. RA = rheumatoid arthritis. DDD = defined daily doses. * Anakinra is an interleukin 1 receptor antagonist that is classified as a “biological response modifier”; its use and expenditure has been included in the total bDMARD and non-anti-TNF bDMARD representations above.
Box 3 –
Use of individual bDMARDs for treating patients with RA in Australia, 2003–2014*
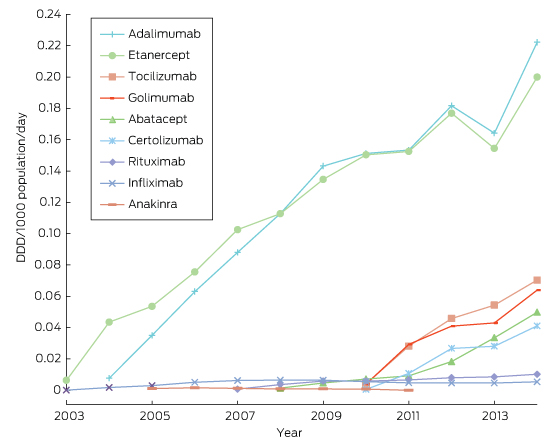
bDMARD = biological disease-modifying antirheumatic drug. RA = rheumatoid arthritis. * Anakinra is an interleukin 1 receptor antagonist that is classified as a “biological response modifier”.
Box 4 –
Cost of various bDMARDs for treating patients with RA in 2014
|
bDMARD |
Typical maintenance dose |
Most dispensed item code (strength) |
Dispensed price for maximum quantity* |
Total dispensing number |
Total expenditure |
Cost per dispensing |
|||||||||
|
|
|||||||||||||||
|
Abatacept |
500–1000 mg IV every 4 weeks (weight based) or 125 mg SC once a week |
1221G (4 × 125 mg) |
$1754 |
15 785 |
$27 559 020 |
$1746 |
|||||||||
|
Rituximab† |
1 g IV for 2-dose course; repeat after > 6 months |
9544H (1 × 500 mg) |
$2033 |
1847 |
$8 296 451 |
$4492 |
|||||||||
|
Tocilizumab† |
4–8 mg/kg IV every 4 weeks (maximum, 800 mg) |
9673D (1 × 400 mg) |
$979 |
12 043 |
$14 753 071 |
$1225 |
|||||||||
|
Adalimumab |
40 mg SC every 2 weeks |
9100Y (2 × 40 mg) |
$1775 |
43 820 |
$77 131 559 |
$1760 |
|||||||||
|
Certolizumab pegol |
200 mg SC every 2 weeks |
3425G (2 × 200 mg) |
$1709 |
12 330 |
$20 927 569 |
$1697 |
|||||||||
|
Etanercept |
50 mg SC once a week or 25 mg twice a week |
9460X (4 × 50 mg) |
$1775 |
31 001 |
$54 565 851 |
$1760 |
|||||||||
|
Golimumab |
50 mg SC once a month |
3429L (1 × 50 mg) |
$1778 |
9851 |
$17 346 793 |
$1761 |
|||||||||
|
Infliximab† |
3–7.5 mg/kg IV every 8 weeks |
5757B (1 × 100 mg) |
$752 |
1041 |
$2 426 802 |
$2331 |
|||||||||
|
|
|||||||||||||||
|
bDMARD = biological disease-modifying antirheumatic drug. RA = rheumatoid arthritis. IV = intravenous. SC = subcutaneous. ∗ Illustrates price to pharmacist for maximum quantity supplied; additional agreed fees may apply.6 † Although the costs of rituximab, infliximab and tocilizumab appear to differ, like other biological agents they have been added to the PBS on a cost minimisation basis, and therefore apparent cost differences are likely a reflection of different administration frequencies or the need for loading doses.5 |
|||||||||||||||
Box 5 –
Criteria for PBS-subsidised initiation of a bDMARD for patients with RA
|
2003 to August 2010 |
August 2010 onwards |
||||||||||||||
|
|
|||||||||||||||
|
Demonstrably severe active RA* that has failed to achieve adequate response to:
|
Demonstrably severe active RA* that has failed to achieve adequate response to:
|
||||||||||||||
|
|
|||||||||||||||
|
PBS = Pharmaceutical Benefits Scheme. bDMARD = biological disease-modifying antirheumatic drug. RA = rheumatoid arthritis. DMARD = disease-modifying antirheumatic drug. ∗ Based on number of joints that are both swollen and tender (> 20 in total or four large joints) and one or more elevated inflammatory markers (erythrocyte sedimentation rate > 25 mm/hour or C-reactive protein level > 15 mmol/L). |
|||||||||||||||
Box 6 –
Service cost per year (combined cost to PBS and RPBS) to provide abatacept, tocilizumab, certolizumab pegol and golimumab to patients with RA, and predicted cost per year based on 5 years of use in RA, 2008–2014
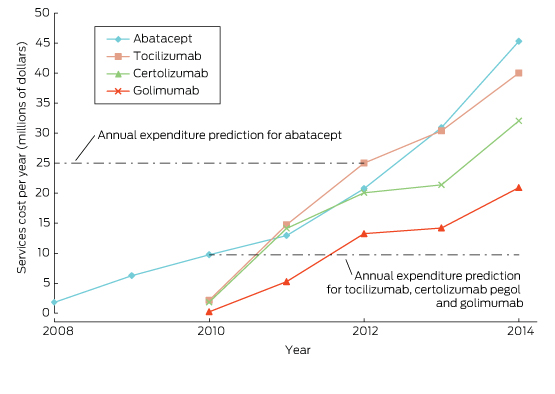
PBS = Pharmaceutical Benefits Scheme. RPBS = Repatriation Pharmaceutical Benefits Scheme. RA = rheumatoid arthritis.
Box 7 –
Example questions to be addressed by a real-world database of restriction criteria data
|
Questions |
|||||||||||||||
|
|
|||||||||||||||
|
What is the response to each bDMARD when prescribed according to current restriction criteria? Do any factors explain variance among patient populations? |
|||||||||||||||
|
Is the cost-effectiveness of bDMARDs, prescribed according to current restriction criteria, comparable with that predicted from trial data? |
|||||||||||||||
|
How do access and response to bDMARDs in remote areas compare with access and response in other areas? What is the differential between private versus public sector access and response? |
|||||||||||||||
|
How many patients have switched to the newer bDMARDs and is this due to documented failure? |
|||||||||||||||
|
What concomitant treatments are patients on bDMARDs receiving (eg, corticosteroids, conventional DMARDs), and do they affect response rates? |
|||||||||||||||
|
Are the response rates for biosimilars equivalent to that for the originator? |
|||||||||||||||
|
What is the impact on immunogenicity of changing between an originator and multiple biosimilar bDMARDs? |
|||||||||||||||
|
|
|||||||||||||||
|
bDMARD = biological disease-modifying antirheumatic drug. DMARD = disease-modifying antirheumatic drug. |
|||||||||||||||