Most of the inflammatory rheumatic diseases are systemic conditions with clinical and pathological manifestations outside of the joints. Many of the extra-articular manifestations of inflammatory rheumatic disease respond to the same treatments that target the joint disease itself, but some require specialised interventions. Ocular involvement is a common manifestation of inflammatory rheumatic disease and ranges from chronic troublesome symptoms, such as dry eye complicating Sjögren syndrome, to organ- and sight-threatening vasculitis. Isolated ocular abnormality has been reported to account for up to 4% of referrals to rheumatology clinics.1 Diagnosing and characterising serious ocular abnormalities are complicated by the need for specialised equipment and training — notably slit lamp and retinal examination — to fully assess eye disease. Consequently, it is incumbent on physicians involved in the care of patients with rheumatic disease to be aware of the potential ocular complications of their patients’ condition and the indications for rapid escalation of treatment to specialist level to preserve sight.
Here, we summarise these important conditions, in order of urgency of treatment, for non-ophthalmologists who are involved in the management of patients with rheumatic diseases (Appendix 1). We identified articles for inclusion in this review by keyword searches in MEDLINE, with an emphasis on up-to-date review articles, and through expert opinion.
Ocular complications of rheumatic diseases
Ischaemic optic neuropathy
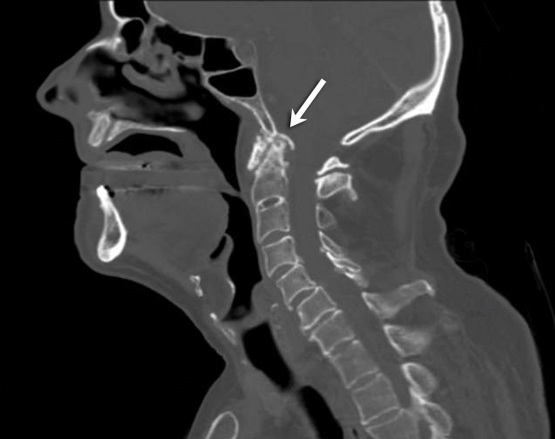
Ischaemic optic neuropathy refers to damage to the optic nerve caused by a lack of blood supply. It is one of the major causes of blindness in older people in the Western world. The systemic disease most often associated with ischaemic optic neuropathy is giant cell arteritis (GCA), in which arterial vasculitis most commonly affects the blood supply to the optic nerve head. This entity is termed arteritic anterior ischaemic optic neuropathy (AAION). About 22% of patients with GCA will experience visual symptoms, with 12% developing irreversible loss of vision.2
GCA, which is by far the most common cause of AAION, rarely affects patients under the age of 50 years and has a mean age at diagnosis of 72 years.3 Women with GCA develop AAION more often than do men (71% v 29%).4 Patients often present with anorexia and the systemic symptoms associated with GCA, such as a persistent temporal headache, scalp tenderness and jaw or tongue claudication. Definitive diagnosis of GCA is made by temporal artery biopsy. The symptoms associated with a positive biopsy result include jaw claudication, neck pain and anorexia.5 Importantly, up to 21% of patients who develop permanent visual loss due to confirmed GCA have no systemic symptoms.5 The visual manifestations of ischaemic optic neuropathy are characterised by painless, sudden loss of vision. Amaurosis fugax is seen in up to 30% of patients with GCA and is a sign of impending loss of vision.4 Inflammatory marker levels are elevated in most patients with GCA, but there is a small proportion of patients with biopsy-proven GCA whose levels are within the normal reference interval.6
Initiation of steroid therapy should not be delayed while awaiting a definitive diagnosis. Treatment of patients with suspected GCA is oral prednisolone (1 mg/kg/day; maximum dose, 60 mg daily) or intravenous (IV) methylprednisolone therapy. A same day referral to an ophthalmologist should be made to arrange biopsy of the superficial temporal artery. The development of amaurosis fugax or any symptoms of visual loss is an indication for pulse IV methylprednisolone therapy.7 Once GCA is confirmed by biopsy, long term oral prednisolone and regular monitoring for systemic side effects of steroids are indicated.
Retinal vasculitis
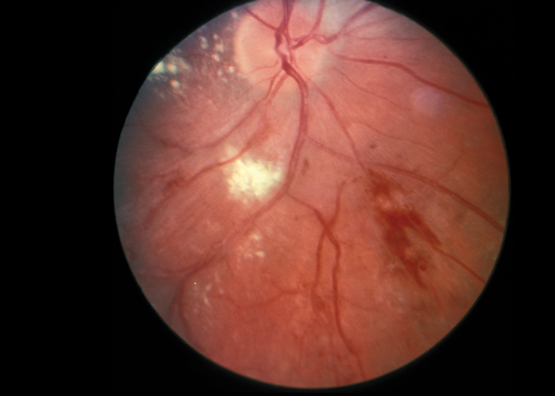
Retinal vasculitis refers to inflammation of the retinal blood vessels. The development of retinal vasculitis is most commonly reported in patients with systemic lupus erythematosus or Behçet disease. Patients who develop retinal vasculitis often progress from mild retinopathy, involving cotton wool spots, perivascular hard exudates, retinal haemorrhages and vascular tortuosity (Box 1), to complete occlusion of retinal arterioles and consequent retinal infarction. Most patients with retinal vasculitis develop proliferative retinopathy, often progressing to vitreous haemorrhage and retinal detachment.8 Evidence of mild retinopathy is seen in 10% of patients with systemic lupus erythematosus8 and up to 74% of patients with the ocular form of Behçet disease.9 Permanent loss of vision is common in those patients who progress to advanced retinal vasculitis.
Retinal vasculitis typically presents with a painless decline in vision. Patients may develop a blind spot from ischaemia-induced scotomas or floaters from vitritis, and those with macular involvement may develop metamorphopsia. It is also possible for retinal vasculitis to be asymptomatic.
Same day referral to an ophthalmologist is recommended for all patients with a painless decline in vision. The diagnosis is often confirmed clinically on detailed retinal examination or with the aid of fluorescein angiography. Aggressive treatment with oral prednisolone (1 mg/kg/day; maximum dose, 60 mg daily) is recommended, often supplemented or replaced with other steroid-sparing immunosuppressive agents. Complications such as proliferative retinopathy are later treated with laser therapy.8
Peripheral ulcerative keratitis
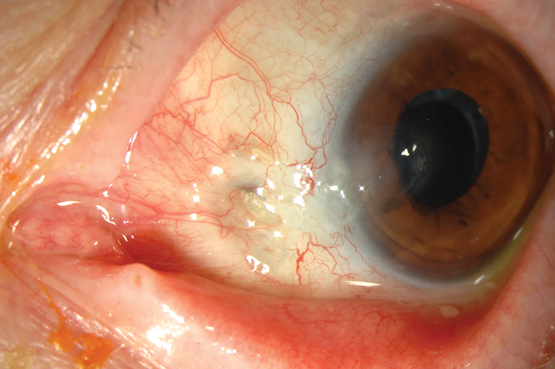
Peripheral ulcerative keratitis (PUK) refers to inflammation and ulceration of the cornea. It is a rare complication of systemic diseases that is characterised by thinning of the stromal layer of the cornea close to the limbus. It is caused by inflammation of the cornea’s stromal layer and is often unilateral and crescent-shaped in appearance. There is an associated epithelial defect overlying the area of inflammation and progressive corneal thinning, which can lead to corneal perforation.10
The development of PUK is most often associated with rheumatoid arthritis but is also reported in cases of polyarteritis nodosa and granulomatosis with polyangiitis. The introduction of biological therapy for rheumatoid arthritis has seen a reduction in the incidence of corneal complications of this disease.11 PUK is reported to have a prevalence in patients with rheumatoid arthritis of 1.4–2.5%.11,12 It is seen most often in patients with longstanding rheumatoid arthritis who are both rheumatoid factor and anticyclic citrullinated peptide antibody positive. The presence of acute anterior uveitis (AAU) and dry eyes is common in patients with PUK.11
Patients with PUK often present with symptoms of ocular redness and pain, tearing, photophobia and decreased visual acuity. As the disease progresses, crescent-shaped corneal ulcers develop, with an associated corneal epithelial defect (Box 2). Inflammation of the surrounding conjunctiva and sclera are frequently apparent.10 Corneal ulceration often abates after treatment, but corneal thinning, scarring and neovascularisation are irreversible.10
The treatment of PUK is determined by the severity of findings within the cornea and the likelihood of imminent perforation. Aggressive treatment of the underlying systemic disease is important to control the progression of corneal disease, and should include a combination of high-dose systemic prednisolone (1 mg/kg/day; maximum dose, 60 mg daily) and an immunomodulatory agent appropriate to the underlying systemic disease, such as methotrexate. Pulse methylprednisolone therapy may be administered in cases of imminent perforation. Surgical management may be required to maintain globe integrity.10
Scleritis
The term scleritis refers to chronic inflammation of the sclera, the dense external covering of the eye. About 25–50% of patients presenting with scleritis have an associated systemic disease,13 most often rheumatoid arthritis (10–18.6%), although scleritis is also seen in patients with granulomatosis with polyangiitis (3.8–8.1%), relapsing polychondritis (1.6–6.4%) and inflammatory bowel disease (2.1–4.1%).14 Scleritis is an important clinical entity to detect, not only because it may lead to loss of vision due to structural changes to the globe but also because it is associated with an increased mortality rate in patients with rheumatoid arthritis.14 Scleritis may involve the anterior sclera, posterior sclera or both, and may be diffuse, nodular or necrotising. Non-necrotising scleritis rarely results in loss of vision, unless it is complicated by uveitis or keratitis. Necrotising scleritis results in rapid thinning of the sclera and exposure of the underlying uvea and is associated with poorly controlled systemic disease. This occurs due to severe vasculitis and closure of the episcleral vascular bed, leading to a visible area of scleral non-perfusion, infarction and necrosis (Box 3).14
Patients with scleritis present with severe pain involving the eye and orbit that radiates to the ear, scalp, face and jaw. The pain is often described as a dull, boring pain that wakes the patient from sleep and is exacerbated by eye movement. Episodes of scleritis can last several months, with the pain increasing in severity over several weeks. Patients with anterior scleritis often notice redness and tenderness of the globe. Patients with necrotising scleritis can experience extreme scleral tenderness. Inflammation often spreads to the surrounding structures, leading to keratitis, anterior uveitis and elevated intraocular pressures, all of which threaten vision.15
Patients with necrotising scleritis require treatment with systemic steroids, either orally or intravenously. A typical starting dose of prednisolone is 1 mg/kg/day (maximum dose, 60 mg daily), which is weaned over the following months based on disease progression. IV methylprednisolone is usually reserved for patients with impending scleral or corneal perforation. Patients who relapse at prednisolone doses of greater than 7.5 mg daily should be considered for adjunctive immunosuppressive therapy, such as methotrexate. Steroid therapy is combined with oral non-steroidal anti-inflammatory agents. Surgical intervention is rarely required in cases of scleral or corneal perforation, which can often be managed with contact lenses and tissue adhesive.15
Uveitis
Uveitis is inflammation of the uvea, the layer of the eye between the outer sclera and the inner retina. It is the most frequently encountered ocular manifestation of rheumatic disease and is responsible for up to 10% of cases of blindness in Western countries.16 Uveitis is classified as anterior, intermediate, posterior or panuveitis, based on the anatomical structures that are inflamed. It is further subdivided into acute, recurrent or chronic, based on the time course of the disease.17
AAU represents the majority of cases of uveitis in patients with rheumatic diseases. This condition involves a sudden onset episode of inflammation of the iris or the anterior ciliary body that is present for less than 3 months.17 Ankylosing spondylitis is the most frequent underlying rheumatic disease causing uveitis, occurring in 20–30% of patients.17 AAU is also associated with other disease entities, such as reactive arthritis, inflammatory bowel disease and psoriatic arthritis. Patients with systemic disease affected by AAU are generally aged between 30 and 40 years, and most test positive for human leukocyte antigen-B27.18
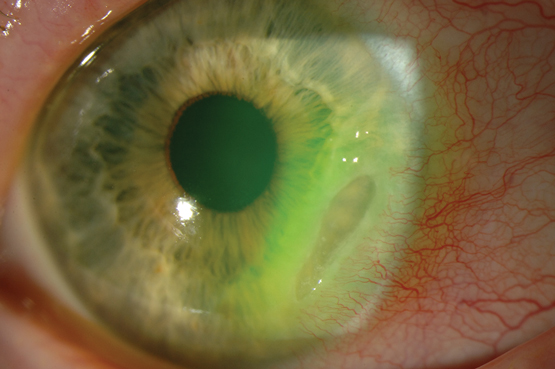
Patients with AAU often present with ocular pain, an acute onset of photophobia, excessive lacrimation, ocular injection and blurred vision (Box 4, Box 5). The symptoms tend to be unilateral and recurrent, occasionally in the contralateral eye. Attacks can last 2–3 months; residual visual impairment is uncommon when treated early.19 Loss of vision in patients with AAU develops as a result of complications, including cataract formation (7–28%),18 macular oedema (up to 11%),20 secondary glaucoma and the development of chronic uveitis.19
AAU is best diagnosed with slit lamp examination, warranting prompt referral to an ophthalmologist when the diagnosis is suspected. The pupil may be poorly reactive and the anterior chamber may show signs of inflammation, including the presence of cells, flare and occasional fibrin (inflammatory debris) within the chamber. In severe cases, there may be evidence of hypopyon. The ciliary blood vessels surrounding the limbus are usually dilated.
The mainstay of treatment is topical glucocorticoids, such as 1% prednisolone acetate. The frequency of the dose depends on the intensity of inflammation present, determined by slit lamp examination.17 Dilating drops are used to relieve pain and prevent iris–lens adhesions.
Sjögren syndrome
Sjögren syndrome is a slowly progressing, immune-mediated inflammatory disease targeting the exocrine glands. It leads to the replacement of functional epithelium with lymphocytic infiltrates, resulting in a dry mouth and dry eyes. Sjögren syndrome is classified as primary when it occurs in the absence of other connective tissue or autoimmune disease, or secondary when it accompanies another disease. About 60% of cases are secondary. It affects women more than men (9:1 ratio), often in their fourth or fifth decade of life.21 Rheumatoid arthritis is the most common underlying rheumatic disease, affecting 10–33% of patients,22,23 followed by systemic lupus erythematosus, affecting up to 9.2% of patients.24 Associations have also been reported in patients with scleroderma and polymyositis.25
The clinical features of Sjögren syndrome are dry eyes and mouth. The discomfort of dry mouth is often associated with difficulty swallowing and speaking, while the lack of tears leads to damage to the conjunctival epithelium over the cornea and globe. This typically presents with dilation of the conjunctival vessels and an irregular appearance to the cornea. Patients may present with dry, gritty or burning eyes, which should prompt consideration of the diagnosis. The average time between the onset of symptoms and the diagnosis of Sjögren syndrome is 10 years,26 largely because the symptoms are considered minor or vague or because they mimic those of other autoimmune diseases. Diagnosis can be made by the combination of the symptoms of dry eyes and mouth, a positive Schirmer test result and positive autoimmune screening (anti-SSA and anti-SSB) blood test results.27 Further characterisation with salivary gland biopsy can be sought if all the above criteria are not satisfied.27
Early diagnosis and treatment are essential for optimal management of Sjögren syndrome. Treatment comprises symptomatic relief through topical replacement lubrication and anti-inflammatory agents, as well as treatment of the underlying systemic disease where appropriate.28
Ocular side effects of drugs used for treating rheumatic disease
The eyes are highly vascular organs with a relatively small mass, making them particularly susceptible to toxic substances that circulate in the blood stream. The fundamental concepts in managing ocular side effects of drugs used in treating rheumatic disease are recognition of the early signs of eye toxicity, withdrawal of the offending agent and referral to an ophthalmologist. These side effects are summarised in Appendix 2.
Corticosteroids
The most studied ocular complications from corticosteroid use are the induction of cataracts and the increase in ocular pressure causing glaucoma.29 Patients treated with oral prednisolone doses of less than 10 mg per day for 1 year have a negligible chance of developing cataracts. In patients taking oral corticosteroids, the yearly incidence of corticosteroid-induced cataracts ranges from 6.4% to 38.7%.30
Glaucoma is thought to develop in patients taking corticosteroids due to glycosaminoglycan and water accumulation in the trabecular meshwork.29 If intraocular pressure is elevated in patients who recently began steroid therapy, the steroids need to be tapered down as rapidly as possible. Current guidelines from the National Health and Medical Research Council recommend survey for glaucoma through regular eye health checks, particularly in patients older than 50 years who have ongoing steroid use.31
Non-steroidal anti-inflammatory drugs
Ocular side effects from non-steroidal anti-inflammatory drugs are rare. Long term use of indomethacin has been associated with cases of corneal opacities and blurred vision.32 Corneal changes diminish or disappear within 6 months of ceasing the offending agent. Celecoxib has been associated with cases of conjunctivitis and blurred vision that resolve rapidly on ceasing the medication.33
Sulfonamides
Patients taking sulfonamides for rheumatoid arthritis, ulcerative colitis or Crohn’s disease are at an increased risk of developing acute transient myopia and acute angle closure glaucoma. These complications have been reported in up to 3% of patients, and often resolve with supportive treatment and withdrawal of the offending agent.34
Biological drugs
Patients taking abatacept report ocular side effects in less than 1% of cases. These include non-specific symptoms such as blurred vision, eye irritation, allergic conjunctivitis and visual disturbances.33
Antimetabolites
Patients taking methotrexate will have a reduction in ocular side effects when it is combined with regular folate supplements. Methotrexate-related ocular toxicities include periorbital oedema, ocular pain, blurred vision, photophobia, conjunctivitis, blepharitis and decreased reflex tear secretion.33
Bisphosphonates
There have been several reports linking bisphosphonate therapy with ocular inflammation, particularly causing uveitis and scleritis. If inflammatory eye disease develops after starting bisphosphonate therapy, discontinuation of the therapy is recommended.33
Antimalarial agents
Hydroxychloroquine sulfate, which has a far lower incidence of retinal toxicity than older quinolones, is the quinolone agent of choice for treating autoimmune disease. Hydroxychloroquine may cause ocular toxicity, including keratopathy, ciliary body involvement, lens opacities and retinopathy, of which retinopathy is of greatest concern.35 While the incidence of true hydroxychloroquine retinopathy is extremely low, the risk increases to close to 1% after 5–7 years of use.36 As such, annual screening by an ophthalmologist is recommended by the American Academy of Ophthalmology for patients who have been taking hydroxychloroquine or chloroquine for more than 5 years.36 Patients should also have a baseline fundus examination and should be informed of the risk of toxicity.36
Conclusion
The development of ocular symptoms in patients with systemic diseases is common, and it is important for physicians to be aware of which symptoms require immediate intervention and ophthalmologist referral to prevent loss of vision. Insight into the epidemiology, clinical presentation, common complications and basic treatment regimens of these conditions enables clinicians to more rapidly identify them in clinical practice.
Box 1 –
Fundoscopic appearance of retinal vasculitis, showing retinal haemorrhages, cotton wool spots and perivascular hard exudates
Box 2 –
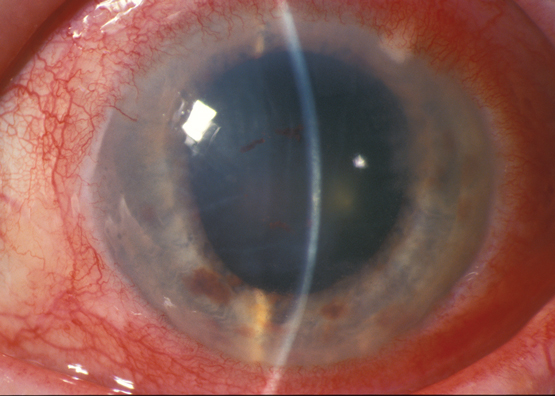
Crescent-shaped corneal ulcer with neovascularisation and injection of ciliary blood vessels
Box 3 –
Necrotising scleritis with exposure of the underlying uvea and associated area of scleral necrosis
Box 4 –
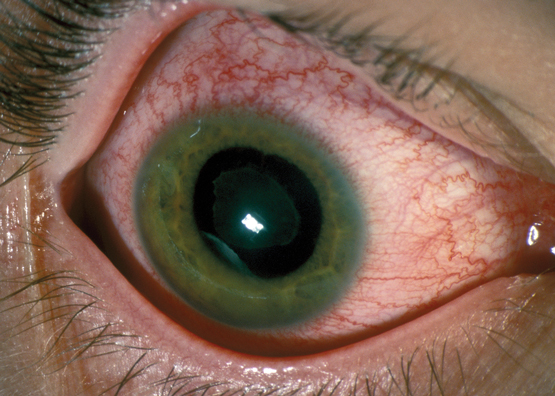
Typical appearance of an eye in a patient with acute anterior uveitis, highlighting the injection of ciliary blood vessels
Box 5 –
Eye of a patient with iritis, showing a red eye with injection of ciliary blood vessels extending to the limbus