Targeted therapy represents a new treatment paradigm that shows great promise in treating serious chronic respiratory diseases for which we presently have limited treatment options.1 Pharmacotherapy in the 20th century was best characterised by the use of antibiotics for infectious diseases. An antimicrobial drug targets a specific pathogen which is the cause of an infectious disease, leading to cure. This approach has its basis in Paul Ehrlich’s magic bullet (zauberkugel) theory and his success with a series of specific antimicrobials for syphilis.2 Although highly effective for acute infectious diseases, this approach cannot be usefully applied to non-communicable chronic diseases (NCCDs) because a single aetiological agent cannot be identified.
Step therapy has emerged as the dominant treatment paradigm for NCCDs such as cardiovascular disease (hypertension, congestive cardiac failure), type 2 diabetes mellitus, cancer and chronic airway disease (asthma, chronic obstructive pulmonary disease). In step therapy, drugs are added (step up) or withdrawn (step down) based on the level of responsiveness to treatment. This has led to improved outcomes but is limited. A key limitation is that individual variability in treatment response is ignored in this one-size-fits-all paradigm.
A new concept is to take individual variability into account when making management decisions. This approach, termed targeted therapy, represents a new treatment paradigm for NCCDs and is defined as “treatments targeted to the needs of individual patients on the basis of genetic, biomarker, phenotypic, or psychosocial characteristics that distinguish a given patient from other patients with similar clinical presentations”.1
In targeted therapy for NCCD, a drug targets a specific pathogenic pathway, leading to major improvements. The corollary is that the treatment is ineffective if the pathway does not operate. This approach is made possible by developments in pathway identification (eg, biomarkers and omics technologies) and specific therapies to target components of pathways (eg, monoclonal antibodies). Targeted therapy is now being successfully applied to chronic respiratory diseases that previously had dismal outcomes. Conditions such as lung cancer, cystic fibrosis (CF) and severe refractory asthma have all seen advances based on the targeted therapy concept. These conditions are associated with premature death, significant quality of life impairment, and a poor response to current therapy. This review highlights these advances (Box 1) using randomised controlled trials published in high impact journals.
Severe asthma
By global standards, Australia suffers a disproportionate burden from asthma. Both the prevalence of and mortality from asthma are among the highest in the world. While major improvements were made in outcomes for mild to moderate asthma in the latter part of the 20th century, these gains have now stalled. Calls for a different approach to asthma care have emerged, and advances have been made in the use of targeted therapy for severe asthma.3
Severe refractory asthma is characterised by severe symptoms, significant lung function impairment and/or asthma exacerbations that occur despite maximal therapy with high dose inhaled corticosteroids and long-acting β-2 agonists.4 It is a high burden disease that affects 3–5% of people with asthma, and up to half of these have eosinophilic disease.5,6 Most patients are unable to work because of their illness,7 and the condition is responsible for almost 50% of direct health care costs for asthma.7 Oral corticosteroids are frequently used to manage severe asthma, however they are only partially effective and are associated with a high prevalence of treatment-related side effects such as type 2 diabetes, osteopenia and osteoporosis, dyspepsia, obesity, hypertension, cataracts, and obstructive sleep apnoea.8
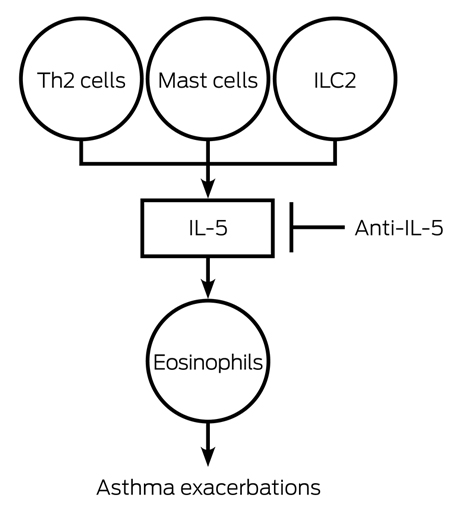
The pathogenesis of severe asthma is heterogeneous. One endotype — severe refractory asthma with eosinophilia (SREA) — is associated with an interleukin (IL)-5 mediated eosinophil influx into the blood and airway (Box 2). Other endotypes include severe refractory allergic asthma, which can be treated with the anti-IgE monoclonal antibody omalizumab, and non-eosinophilic asthma, for which targeted therapy is currently lacking. IL-5 plays a fundamental role in eosinophil growth, differentiation and survival. Human eosinophils express comparatively high levels of IL-5 receptor subunit alpha compared with other cells. The major cellular sources of IL-5 in asthma are T helper type 2 (Th2) cells, mast cells, group 2 innate lymphoid cells, and eosinophils themselves. Monoclonal antibodies directed against IL-5 (mepolizumab, reslizumab) or its receptor (benralizumab) have been developed to treat SREA. Studies consistently show that mepolizumab reduces blood and airway eosinophils in asthma.9
The clinical efficacy of mepolizumab has now been demonstrated in several randomised controlled trials (RCTs). The key effects are a highly significant reduction in asthma exacerbations10,11and an oral steroid-sparing effect.12 Some studies also show improvement in lung function and quality of life.11 These trials recruited adults and adolescents with severe asthma, persistent asthma exacerbations despite inhaled therapy, and evidence of eosinophilia from blood eosinophil counts > 0.3 cells × 109/L, sputum eosinophil counts ≥ 3%, or an elevated fraction of exhaled nitric oxide (a marker of Th2-mediated inflammation). The DREAM study10 was a dose-ranging, parallel group, multicentre, double-blind RCT of mepolizumab in which 621 participants were randomised to one of three doses of intravenous mepolizumab or placebo every 4 weeks for a year. The results showed a highly significant reduction in asthma exacerbations between 39% and 52%, which have been replicated in subsequent RCTs.11,12 Mepolizumab treatment of patients with severe oral corticosteroid-dependent asthma was associated with a median 50% reduction in maintenance corticosteroid dose.12 The safety profile was similar to placebo. Mepolizumab is licensed for use by the United States Food and Drug Administration (FDA) and has been approved in Australia by the Therapeutic Goods Administration (TGA) but is not funded for clinical use at this time.
The studies show preferential effects of anti-eosinophil therapy on asthma exacerbations consistent with studies that target airway eosinophilia in severe asthma.13 The level of blood eosinophils used to select participants for targeted therapy in severe asthma is within the quoted normal range; however, studies of normal values that exclude people with atopic disorders have found a lower level for the upper normal range.14 Values of blood eosinophils > 0.3 cells × 109/L have been associated with airway eosinophilia in corticosteroid-treated asthma.15
Therapy targeted at the IL-5/eosinophil endotype of asthma is highly effective. In adults and adolescents with severe refractory eosinophilic asthma recognised by a persistence of blood eosinophils despite corticosteroid therapy, the addition of targeted therapy against IL-5 or its receptor may reduce asthma exacerbations by almost 50% and reduce oral corticosteroid requirements. Administration and access require a systematic assessment of asthma and management of potentially reversible factors, including non-adherence to maintenance corticosteroid therapy.
Cystic fibrosis
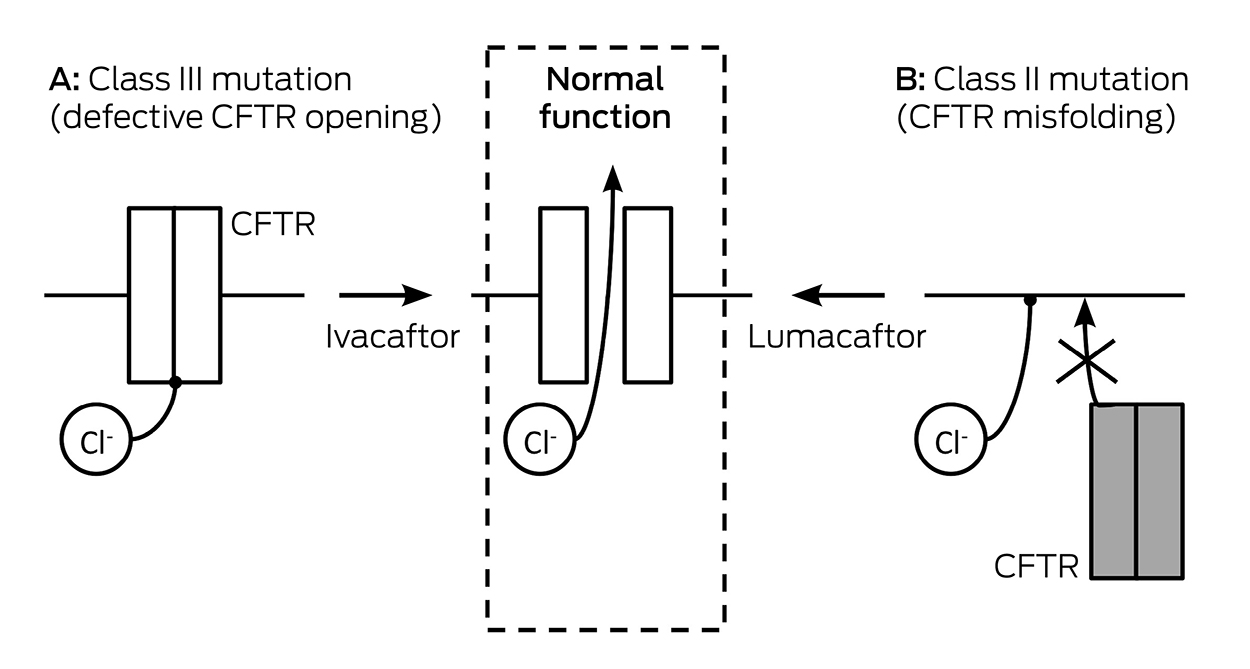
CF is a multisystem genetic disorder that primarily affects the lungs causing bronchiectasis, recurrent infections, and premature death. In CF, there is a defective or deficient CF transmembrane conductance regulator (CFTR) protein that functions as an anion channel predominantly in epithelial cell membranes. There are about 3300 patients with CF in Australia, most of whom are adults. The CFTR gene was discovered in 198916 and has over 2000 different mutations17 that may cause either an inadequate amount of CFTR protein or deficient function of CFTR protein at the epithelial cell membrane, or a combination of these abnormalities (Box 3). Deficient function may be due to a reduced open probability of the CFTR channel (gating) or abnormal conductance of the channel. The most common CFTR mutation is Phe508del (formerly F508del); about 45% of patients are homozygous for this class II mutation worldwide.18 Therapies targeted at specific defects in CFTR function have been developed, and new therapies are currently in development.
In 2004, ivacaftor, the first CFTR modulator approved for clinical use, commenced development. Ivacaftor targets the deficient function of CFTR protein at the epithelial membrane and increases the open probability (gating) of the CFTR channel. It is referred to as a CFTR potentiator (Box 3). The most common CFTR gating mutation is Gly551Asp (formerly G551D), a class III mutation which accounts for about 4% of CFTR alleles; it is associated with an adequate amount of protein at the epithelial membrane but the protein has no CFTR function. Two landmark phase 3 trials of ivacaftor in patients aged 6–11 years and 12 years and older carrying at least one copy of the Gly551Asp mutation19,20 demonstrated improvements in lung function (mean absolute improvement in the percentage of predicted forced expiratory volume in 1 second [FEV1] of about 10.6%) and nutritional status, and reported a marked reduction in sweat chloride measurement confirming the effect of restoration of CFTR function. These clinical benefits persisted in a 2-year open-label extension study.21 In 2012, the US FDA approved the use of ivacaftor for patients aged 6 years and older with CF carrying at least one Gly551Asp mutation. Successful clinical trials of ivacaftor permitted extension of approval to eight other non-Gly551Asp gating mutations,22 and to patients carrying a residual function Arg117His (formerly R117H) mutation.23 In Australia, ivacaftor is Pharmaceutical Benefits Scheme (PBS)-listed on the high cost drug program for patients aged 6 years and older with CF carrying at least one Gly551Asp mutation or one of the other eight gating mutations.
The Phe508del mutation results in a processing defect with almost no protein available at the epithelial membrane. Where small amounts of the protein are present or can be rescued, it nonetheless has a gating defect and is unstable and more rapidly cleared. A combination of therapies aimed at both rescuing and potentiating Phe508del was therefore considered for patients with CF and the Phe508del mutation. Lumacaftor, a CFTR corrector that targets class II defects, was shown in vitro to correct p.Phe508del CFTR misprocessing and increase the amount of available protein.24 Ivacaftor was shown in vitro to potentiate p.Phe508del,25 and the combination of the two drugs in vitro resulted in greater chloride transport than either drug alone.24 The in vitro experience was confirmed in clinical trials, as monotherapy with each of these agents was found not to have clinical benefit.26,27 Combination therapy with lumacaftor and ivacaftor was shown in a large phase 3 trial in patients aged 12 years and older with CF and who were homozygous for the Phe508del mutation to provide modest improvement in lung function (mean absolute improvement in FEV1, 2.6–4%) and reduce pulmonary exacerbations as well as provide nutritional benefit.28 Combination therapy with lumacaftor and ivacaftor was approved by the FDA in July 2015 for patients aged 12 years and older who are homozygous for Phe508del. In Australia, lumacaftor–ivacaftor combination therapy has been approved by the TGA but is not funded for clinical use at this time.
There has been less success to date with CFTR modulator therapies for class 1 mutations,29 and no drugs are currently approved for patients with such mutations. The targeted therapy approach has shown that it is possible to correct the underlying defect in some patients with CF and achieve clinically important benefit.30 Specific approaches are required for different CFTR mutations and differing clinical benefits have been observed. The potential for long term disease modification over time and in early life will need to be taken into account when considering the overall clinical and health economic benefit of these therapies. In addition, maintaining optimal outcomes from the use of ivacaftor, lumacaftor–ivacaftor combination or other CFTR modulator therapies as they become available in clinical use will require careful assessment of patient CF genotype and assessment of the potential for clinical benefit as well as education and support of patients and families to ensure these medicines are taken correctly and appropriately with long term monitoring for adverse events and drug interactions.
Lung cancer
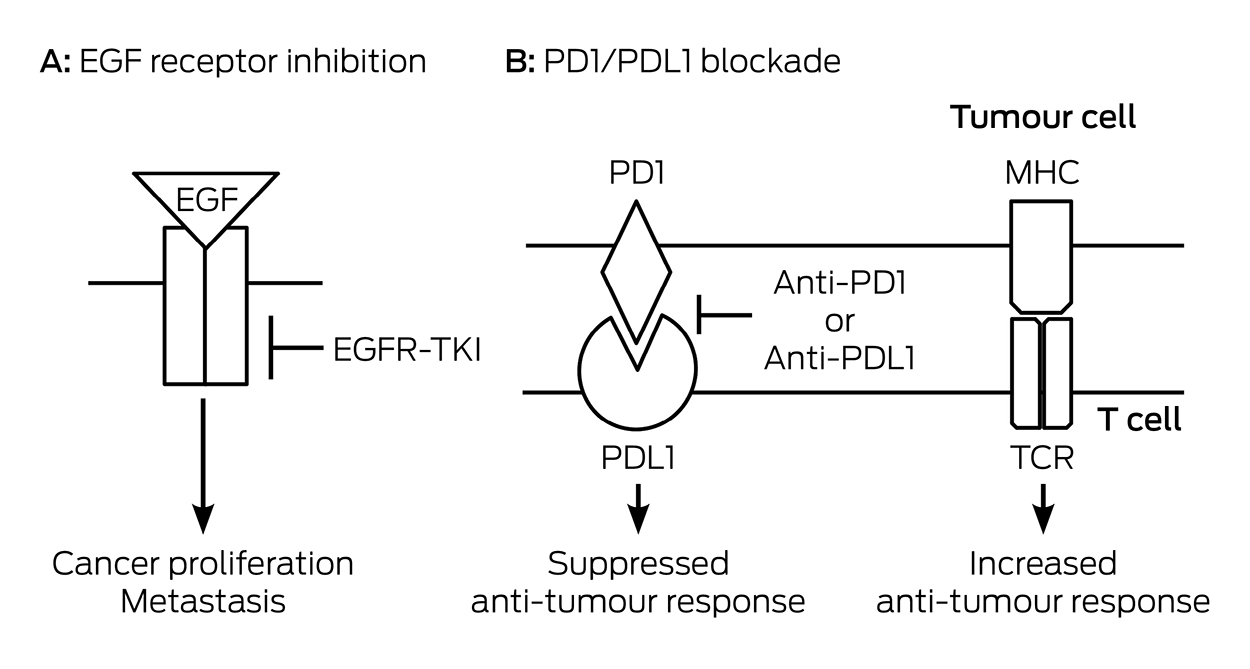
Lung cancer is the most common cause of cancer mortality in Australia, with nearly 9500 deaths annually.31 Tumours with similar pathological appearance may have different genetic abnormalities. Specific mutations can be used to guide optimal treatment. In lung cancer, epidermal growth factor (EGF) binds to its specific receptor causing tumour growth. In early trials that recruited unselected patients, EGF receptor tyrosine kinase inhibitors (EGFR-TKIs) gefitinib and erlotinib were disappointing until it was recognised that the dramatic responses seen in a subset of patients32 were in patients whose tumours carried specific EGFR gene mutations: deletions within exon 19 or a specific point mutation in exon 21. EGFR-TKI treatment is superior to cytotoxic chemotherapy only when these specific mutations are present (Box 4). EGFR mutation testing should be performed on all advanced non-squamous non-small cell lung cancer (NSCLC). This requires larger tissue biopsy33,34 but expertise to meet this need exists and blood testing for tumour DNA may soon be available. Typical time before progressive disease emerges is 2–3 years, which well exceeds that of previously standard cytotoxic chemotherapy.
Additional targets have been identified such as the EML4-ALK and ROS-1 fusion genes. Crizotinib is an available targeted treatment with a high response rate for EML4-ALK-mutated tumours. It is also effective for tumours that carry a ROS-1 fusion gene but it is not presently subsidised under the PBS for this purpose. Tumour progression after a good initial response to EGFR-TKIs and EML4-ALK inhibitors is eventually seen. Newer agents such as osmertinib in the case of EGFR mutations and alectinib for EML4-ALK mutations are effective for the common additional resistance mechanisms but none is presently available via the PBS.35 These oral targeted treatments have side effects and a treatment benefit versus toxicity trade-off must be made. A distinctive rash is common with EGFR-TKIs and should be aggressively managed. Crizotinib has gastrointestinal effects and some visual symptoms are common. Adverse effects are less than with cytotoxic chemotherapy but may not be trivial and persist for as long as treatment continues.
Immune checkpoint inhibitors are a class of treatments with similar promise (Box 4). They prevent interactions between the programmed death 1 (PD1) protein and its ligand that suppress host versus tumour immunity. Unlike the tumour mutations described, PD1 expression is not a great predictor of the effectiveness of such treatments. The response rate is not as high as with EGFR-TKIs but there may be more enduring responses.
Between 500 and 1000 Australians annually may be suitable for current targeted treatment — more than the total incidence of many common cancers. If second line TKI treatments and checkpoint inhibitors are as effective as initial data suggest, careful consideration of cost, benefit and equity of access will be required. Oral treatments do provide some offset by reducing day-stay infusion costs. Refusal of funding for checkpoint inhibitors will be ethically challenging if the effect is as great in lung cancer as it is in melanoma, where access is already provided.
Related issues
Study design
Targeted therapy raises several issues (Box 5) regarding study design that require a re-examination of the traditional parallel group RCT used to establish drug efficacy. Large parallel group RCTs use representative samples of the population in order to test the efficacy of a therapy. However, targeted treatments are only effective in a subset of patients, specifically those in whom the targeted pathway or mutation is active. This means that accurate subject selection is a key part of the evaluation of a targeted treatment, and failure to do this can result in the rejection of a potentially effective therapy. There are examples of this in both severe asthma and NSCLC trials. A study of mepolizumab in unselected asthma patients was negative,36 indicating that while the targeted therapy may be highly effective in patients with an active eosinophil pathway,10–12 it may not be effective in those without this pathway operating. Similarly, initial trials of EGFR-TKIs involving unselected populations with NSCLC found no effect on survival.37 Consequently, privileging RCTs when trying to assess the effects of targeted treatment has been called into question.38
A further study design problem that occurs with targeted therapy relates to clinical trial access. Trials are more challenging in infants and young children, patients with more severe illness and patients with rarer disease subtypes, such as those involving rarer CFTR mutations. New approaches will be needed to ensure equitable access to new therapies for all patients with diseases undergoing a targeted therapy approach. As more patients start to use targeted therapy, the pool of patients naive to targeted treatment will decline, which will make placebo-controlled trials less possible. Comparative effectiveness studies will likely be used.
Maintaining adherence remains a challenge for all therapies. For example, CFTR modulator therapies require oral administration with fatty food to maximise absorption and adherence can be suboptimal.39 Patients may also feel healthier taking targeted therapies and there is a risk of reduced adherence with other maintenance therapies. How this may affect the long term benefits of treatment is unknown, although the existing clinical trials of targeted therapy have been undertaken with careful support of patients to maintain their therapeutic regimens.
Cost
The financial costs of currently available targeted therapies are high, with costs estimated at up to $300 000 per patient per year for some therapies. Currently, relatively small numbers of patients with lung cancer, CF or severe asthma benefit from the personalised medicines available. As a society, we will need to discuss and plan how we can manage the costs of personalised medicine into the future to enable equitable access as new medicines become available and the overall numbers of patients using highly expensive medicines grow. Australia has developed a framework to address aspects of this developing treatment paradigm.40
Therapies are currently approved based on an average improvement in a clinical outcome, yet in reality they only help a small proportion of the people who receive them. For example, the top ten highest-grossing drugs in the US help between one in 25 and one in four people who use them.41 This has led to calls for greater implementation of personalised medicine as a means to better use resources based on nationally consistent collection of data.1,3,38
Conclusion
Targeted therapy is a new treatment paradigm that has demonstrated improved efficacy for serious chronic respiratory diseases such as severe asthma, CF and lung cancer. Implementing targeted therapy offers hope to many patients with chronic respiratory diseases but also creates challenges for study design and equitable delivery of new medications. Health economic considerations require that clinicians cease the therapy if the expected response is not seen in individual patients.
Box 1 –
Targeted therapies for chronic respiratory diseases
|
Disease
|
Target
|
Therapy
|
Biomarker
|
Bio-effect
|
Clinical effect
|
|
|
Severe asthma
|
IL-5
|
Mepolizumab; reslizumab
|
Eosinophils (blood)
|
Reduced eosinophils
|
Reduced asthma exacerbations
|
|
Cystic fibrosis
|
CFTR
|
Ivacaftor
|
CFTR gating mutations: eg, Gly551Asp
|
Reduced sweat chloride
|
Increased lung function, improved nutrition and reduced exacerbations
|
|
|
|
Lumacaftor–ivacaftor combination
|
Phe508del-CFTR mutation
|
Reduced sweat chloride
|
Increased lung function, improved nutrition and reduced exacerbations
|
|
Lung cancer
|
EGFR-TKI; EML4-ALK; ROS-1
|
Gefitinib; erlotinib; crizotinib*
|
EGFR mutation; gene fusion
|
Tumour regression
|
Increased progression-free survival
|
|
|
IL-5 = interleukin-5. CFTR = cystic fibrosis transmembrane conductance regulator. EGFR-TKI = epidermal growth factor receptor tyrosine kinase inhibitor. * Crizotinib is approved by the Therapeutic Goods Administration but is not listed on the Pharmaceutical Benefits Scheme.
|
Box 2 –
Targeted therapy in asthma blocks the interleukin (IL)-5 pathway in severe refractory eosinophilic asthma
Box 3 –
CFTR mutational classes and molecular consequences in cystic fibrosis
Box 4 –
Targeted therapy in non-small cell lung cancer
Box 5 –
Targeted medicine: unresolved questions
|
|
- What is the best name for this approach? Targeted therapy, personalised medicine or precision medicine?
- What are the implications for study design, drug regulation, health economics and companion diagnostics?
- Moving beyond targeted pharmacotherapy: can this approach be generalised to problem-based management of chronic disease?
- Translating into practice: what are the best models to implement targeted therapy in practice?
|
|
|
|