Multidrug-resistant tuberculosis (MDR-TB), defined by resistance to both isoniazid and rifampicin, has significant implications for individual patient management and TB control efforts. The current global situation is further complicated by the emergence of extensively drug-resistant TB (XDR-TB), defined by additional resistance to a fluoroquinolone and at least one second-line injectable drug (amikacin, kanamycin or capreomycin).1 Drug resistance may develop in the context of TB treatment, but the majority of MDR-TB cases are contracted as primary infections.2 As with drug-susceptible TB, household transmission is common, frequently affecting young children.3,4 Treatment is resource-intensive and requires longer courses of less effective, more toxic and more expensive drugs compared with drug-susceptible TB.5
Global efforts to combat the threat of MDR-TB have been hampered by a paucity of data. Although progress has been made towards obtaining accurate estimates of MDR-TB in key high-burden countries, less than 4% of bacteriologically proven incident TB cases worldwide underwent formal drug susceptibility testing (DST) in 2011.1 Overall, 3.7% of new TB cases are estimated to be MDR-TB, with proportions by country varying from 0 to 32.3%. The estimated treatment success of MDR-TB globally is 48%.1 Even in wealthy countries, MDR-TB is associated with increased risk of adverse outcomes, including death.6–8
A total of 196 laboratory-confirmed MDR-TB cases were reported in Australia from 1998 to 2010.9 In Victoria, increasing numbers of MDR-TB cases were reported over the 10-year period to 2007.10 Most patients were born overseas, but local transmission has also been reported.11 High rates of MDR-TB (about 25% of tested isolates) have been observed in patients from Papua New Guinea who were treated in Queensland health clinics in the Torres Strait.12 XDR-TB remains rare, with only two reports in Australia.9,13
Early experience of MDR-TB in Western Australia was published in 1991.14 Here, we describe epidemiological, clinical, treatment and outcome data for all MDR-TB cases notified in WA over 15 years to 2012, and compare MDR-TB cases against a matched cohort of patients with drug-susceptible TB.
Methods
All patients with a laboratory-confirmed diagnosis of MDR-TB in WA from 1 January 1998 to 31 December 2012 were identified from the state Mycobacterium Reference Laboratory in Perth. Automated DST was carried out using the BACTEC 460TB mycobacterial detection system (Becton Dickinson) before 2007 and the BACTEC MGIT 960 system (Becton Dickinson) since then. Isoniazid susceptibility was tested at 0.1 μg/mL and 0.4 μg/mL in each case. Paediatric patients with probable MDR-TB, diagnosed according to international research definitions on the basis of probable TB plus a history of household or daily contact with someone with confirmed MDR-TB,15 were also included.
For each MDR-TB case, three matched controls with drug-susceptible TB (on the basis of DST or demonstrated response to standard therapy) were selected from the same period. Randomly chosen controls were matched for site of TB disease, HIV status, age and sex.
De-identified patient data were collected from medical and laboratory records for all cases and controls. Data included demographic characteristics, risk factors, clinical and laboratory diagnostic information, treatment details, health care resource use and outcomes.
Statistical analysis was performed with GraphPad Prism 6.0 statistical software (GraphPad). Categorical data were compared using McNemar’s test, and continuous variables using the Mann–Whitney test. A two-tailed P value < 0.05 was considered significant.
Ethics approval for the study was granted by the WA Department of Health Human Research Ethics Committee.
Results
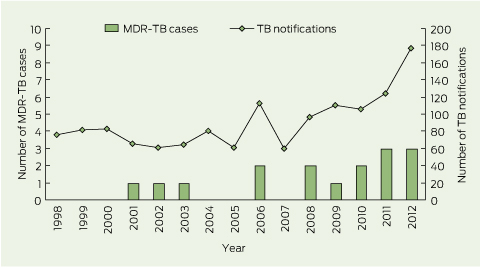
During the study period, 16 cases of MDR-TB were notified (zero to three cases per year), accounting for 1.2% of all TB cases (n = 1352) notified in WA (Box 1). Fifteen cases were laboratory-confirmed MDR-TB. One case was defined as probable MDR-TB on the basis of a clinical syndrome consistent with TB (clinical features, neuroimaging and cerebrospinal fluid examination suggestive of tuberculous meningitis) and a previous isolate of laboratory-confirmed MDR-TB from the same patient.
Patients with MDR-TB were predominantly female (12/16), with a median age of 26 years (range, 8–58 years). Most patients (15/16) were born outside Australia (East Asia and Pacific, 8; sub-Saharan Africa, 4; South Asia, 2; Middle East and North Africa, 1). Refugees with humanitarian visas and asylum seekers in Australian detention centres each accounted for two MDR-TB cases.
Rates of TB risk factors were similar between cases and controls, although patients with MDR-TB were more likely to have been previously treated for TB with a regimen containing rifampicin and isoniazid (Box 2). However, most patients with MDR-TB had never been exposed to antituberculous therapy.
Pulmonary disease was most common (11/16), with positive sputum smear microscopy results noted in about half of pulmonary cases (Box 3). Extrapulmonary manifestations included tuberculous meningitis, genitourinary TB, lymphadenitis and pleural TB. Of the patients who received effective therapy, those with MDR-TB were more likely to experience delays of 1 week or more from specimen collection to commencement of treatment (11/13 [85%] v 14/48 [29%]; P < 0.001).
Of the 15 laboratory-confirmed cases, 13 demonstrated high-level resistance to isoniazid at 0.4 μg/mL. Resistance to ethambutol and pyrazinamide was common. No XDR-TB cases were identified, although resistance to second-line agents including ciprofloxacin or ofloxacin and amikacin was occasionally seen (Box 3).
Hospitalisation was more common for patients with MDR-TB than controls and, for those who completed therapy, their mean duration of treatment was more than twice as long (Box 4). Targeted second-line antituberculous drugs, individualised on the basis of DST, were used in 13 MDR-TB cases. All regimens included moxifloxacin and an injectable agent for at least part of the treatment course; moxifloxacin was ceased in one case shown to be quinolone-resistant.
Adverse effects were more commonly reported in patients with MDR-TB and necessitated modification of therapy in five patients (Box 4). Symptoms reported in patients with MDR-TB but not in those treated for drug-susceptible TB included vestibular toxicity and hearing impairment secondary to injectable aminoglycosides, and neuropsychiatric problems that were attributed to MDR-TB drugs in seven patients (Box 4).
One paediatric patient with laboratory-confirmed pulmonary MDR-TB was treated with isoniazid, rifampicin and pyrazinamide for 12 months, with apparent initial success but subsequent relapse (culture-negative meningitis) 2 years later, which was successfully treated with second-line agents for 24 months. No other treatment failures or deaths occurred in either group, although treatment was ongoing in four MDR-TB patients and three controls at the end of the study period. Three MDR-TB patients and seven controls were transferred out before completion of therapy (Box 4).
Screening for TB infection was carried out for 727 contacts of patients with MDR-TB (median, 6; range, 0–625) and 371 contacts of controls (median, 3; range, 0–222). No secondary cases of active MDR-TB disease were identified.
Discussion
MDR-TB remains uncommon in WA, though the challenges associated with managing it are increasingly recognised. We found that, despite an association with previous TB treatment, most cases occurred through primary transmission. Most patients with MDR-TB diagnosed in WA were born in one of the 27 high MDR-TB burden countries.1
Delayed diagnosis, which has an impact on timely provision of effective therapy and increases the risk of local transmission of MDR-TB strains, is a significant concern.13 Traditional methods for TB culture and DST take several weeks to produce results, contributing to delays. Nucleic acid amplification tests (NAATs), such as the World Health Organization-endorsed Xpert MTB/RIF assay (Cepheid), can rapidly detect TB and the rpoB gene mutation that confers rifampicin resistance.16 We have not reported information about the use of NAATs in this study, as they were only introduced into routine methods in 2011. Caution is warranted in the interpretation of rapid tests for rifampicin resistance due to low positive predictive value when the pretest probability of rifampicin resistance is low.16 Nonetheless, in patients at higher risk of MDR-TB (those with previous TB treatment, a household MDR-TB contact or residence in a high MDR-TB burden country), the use of a rapid NAAT to detect rifampicin resistance may hasten diagnosis. If conducted routinely in a low-prevalence setting, NAAT results should be interpreted cautiously and should be in addition to formal DST.
As is appropriate in a setting with ready access to DST, patients with MDR-TB in WA were managed with individualised drug regimens. Later-generation fluoroquinolones, such as moxifloxacin, are the most potent bactericidal drugs available for the treatment of MDR-TB. Their use has been associated with increased chance of treatment success.17 Moxifloxacin was administered to all 13 MDR-TB patients treated with second-line drugs in this study. Studies have demonstrated improved outcomes with regimens including at least 18 months of active therapy, and the WHO recommends a minimum treatment duration of 20 months for MDR-TB.18–20 Research continues into the possibility of effective shorter-course regimens as brief as 9 months.21 All nine patients in this study who completed an MDR-TB targeted regimen received at least 18 months of active therapy. Pending further research, this conservative approach should be the preferred option in clinical settings where MDR-TB is treated.1,18
Adverse drug reactions more commonly complicate the treatment of MDR-TB than drug-susceptible TB. Close clinical and laboratory follow-up is obligatory for all patients with MDR-TB, and directly observed therapy should be considered where possible. Drugs that are often poorly tolerated, such as prothionamide, cycloserine and para-aminosalicylic acid (PAS), may be initiated gradually.8 Patients receiving aminoglycoside therapy should undergo regular screening for ototoxicity. Cessation of problematic drugs may be unavoidable, as was the case for one patient in WA who experienced severe psychiatric symptoms with unmasking of post-traumatic stress disorder after commencing cycloserine. Unfortunately, alternative options for treatment may be limited.
The complexity and length of MDR-TB treatment necessitates significant health care resource use, placing increased demands on outpatient and inpatient services. Specialist TB services play an important role in the effective management of TB and are crucial for accurate diagnosis and adequate management of protracted MDR-TB treatment regimens and their associated toxicities.
Given the clinical and public health implications of MDR-TB, prevention should be a priority. Prevention of acquired resistance is achieved by ensuring early diagnosis and effective treatment of all TB cases. Prevention of MDR-TB transmission requires early diagnosis, effective treatment and appropriate infection control measures. About a third of patients with MDR-TB in this series were infectious at the time of diagnosis on the basis of positive sputum smear microscopy results. Contact tracing after a new diagnosis of MDR-TB is recognised as an important measure in identifying further cases. This has significant workforce implications. Guidance on management of MDR-TB contacts found to have latent TB infection is currently limited.3,4,11
Our study has several limitations. Comparison of clinical and diagnostic information was affected by inconsistency in diagnostic approach and the use of matched controls. The ability of the study to detect a difference in outcomes was affected by the small numbers analysed. A quarter of patients with MDR-TB were still receiving treatment at the time of data collection. Of the remaining patients, 75% successfully completed treatment, compared with 84% of patients with drug-susceptible TB. In both groups, patients who did not achieve treatment success were transferred out before completion of therapy. While some patients transferred of their own volition, several patients with drug-susceptible TB and one with MDR-TB were deported on the basis of rejected asylum claims. In contrast, consensus recommendations urge that:
All patients with TB who present to health care services within Australia’s borders should have free and equal access to TB care from diagnosis to completion of treatment, irrespective of their legal status or other demographic characteristics …22
In conclusion, MDR-TB is uncommon in WA and is usually associated with treatment success, despite delays to effective therapy and frequent therapeutic changes due to adverse effects. Early diagnosis of MDR-TB is important for both individual patient care and to reduce the risk of transmission. Long treatment courses are associated with increased health service use. Further research into optimal treatment regimens is required. Specialist TB services are heavily relied on for prevention and management of MDR-TB and should be strengthened to effectively control TB and limit the emergence of MDR-TB in Australia and the surrounding region.
1 Multidrug-resistant tuberculosis (MDR-TB) cases and total TB notifications in Western Australia, 1998–2012
2 Risk factors in patients with multidrug-resistant tuberculosis (MDR-TB) and matched controls with drug-susceptible TB in Western Australia, 1998–2012
|
Risk factor
|
MDR-TB (n = 16)
|
Susceptible TB (n = 48)
|
P
|
|
|
Born in a high-prevalence country*
|
15 (94%)
|
39 (81%)
|
0.11
|
|
Resident > 3 months in a high-prevalence country*
|
16 (100%)
|
41 (85%)
|
0.02
|
|
Born in a high MDR-TB burden country†
|
10 (63%)
|
21 (44%)
|
0.07
|
|
Previous TB diagnosis treated with first-line TB drugs
|
4 (25%)
|
1 (2%)
|
0.006
|
|
Previous treatment with isoniazid monotherapy
|
0
|
2 (4%)
|
0.48
|
|
Household TB contact
|
6 (38%)
|
17 (35%)
|
1.0
|
|
Household MDR-TB contact
|
1 (6%)
|
0
|
0.25
|
|
HIV
|
1 (6%)
|
Matched
|
|
|
|
* Country with TB prevalence > 50 per 100 000 population. † One of 27 high MDR-TB burden countries that account for 85% of estimated MDR-TB cases globally.1
|
3 Diagnostic details for patients with multidrug-resistant tuberculosis (MDR-TB) and matched controls with drug-susceptible TB in Western Australia, 1998–2012
|
Diagnostic detail
|
MDR-TB (n = 16)
|
Susceptible TB (n = 48)
|
P
|
|
|
Pulmonary TB
|
11 (69%)
|
Matched
|
|
|
Extrapulmonary TB
|
5 (31%)
|
Matched
|
|
|
Central nervous system
|
1 (6%)
|
|
|
|
Genitourinary
|
1 (6%)
|
|
|
|
Lymph node
|
2 (13%)
|
|
|
|
Pleural
|
1 (6%)
|
|
|
|
Sputum smear microscopy positive for acid-fast bacilli
|
5 (31%)
|
18 (38%)
|
|
|
TB culture positive
|
15 (94%)
|
37 (77%)
|
|
|
Drug resistance
|
|
|
|
|
Isoniazid
|
15/15 (100%)
|
2/37 (5%)
|
|
|
Rifampicin
|
15/15 (100%)
|
0
|
|
|
Ethambutol
|
7/15 (47%)
|
0
|
|
|
Pyrazinamide
|
5/15 (33%)
|
1/37 (3%)
|
|
|
Streptomycin
|
10/15 (67%)
|
4/37 (11%)
|
|
|
Amikacin
|
1/15 (7%)
|
Not tested
|
|
|
Capreomycin
|
1/15 (7%)
|
Not tested
|
|
|
Ciprofloxacin or ofloxacin
|
2/15 (13%)
|
Not tested
|
|
|
Ethionamide
|
3/15 (20%)
|
Not tested
|
|
|
How case was identified
|
|
|
|
|
Contact tracing
|
1 (6%)
|
1 (2%)
|
|
|
Routine screening
|
5 (31%)
|
19 (40%)
|
|
|
Symptomatic presentation
|
10 (63%)
|
28 (58%)
|
|
|
Time to TB notification from arrival in Australia < 1 year
|
6/15 (40%)
|
18/45 (40%)
|
|
|
Delay from specimen collection to effective TB treatment < 1 week
|
2 (13%)
|
34 (71%)
|
0.01
|
|
Never received effective TB treatment
|
3 (19%)
|
0
|
0.008
|
|
Median days of delay for those with ≥ 1-week delay to effective treatment (range)
|
48 (17–149)
|
21 (7–84)
|
0.002
|
4 Treatment details and outcomes for patients with multidrug-resistant tuberculosis (MDR-TB) and matched controls with drug-susceptible TB in Western Australia, 1998–2012
|
Treatment detail/outcome
|
MDR-TB
(n = 16)
|
Susceptible TB
(n = 48)
|
P
|
|
|
Hospitalised during treatment
|
16 (100%)
|
17 (35%)
|
< 0.001
|
|
Mean total days in hospital (range)
|
26 (1–99)
|
13 (2–41)
|
|
|
Directly observed therapy
|
14 (88%)
|
6 (13%)
|
< 0.001
|
|
Intravenous access required for treatment
|
11 (69%)
|
0
|
< 0.001
|
|
Drugs used in definitive treatment regimen
|
|
|
|
|
Isoniazid
|
1 (6%)
|
48 (100%)
|
|
|
Rifampicin
|
1 (6%)
|
48 (100%)
|
|
|
Ethambutol
|
7 (44%)
|
43 (90%)
|
|
|
Pyrazinamide
|
10 (63%)
|
48 (100%)
|
|
|
Moxifloxacin
|
12 (75%)
|
2 (4%)
|
|
|
Prothionamide
|
10 (63%)
|
1 (2%)
|
|
|
Cycloserine
|
10 (63%)
|
0
|
|
|
Amikacin
|
9 (56%)
|
0
|
|
|
Capreomycin
|
2 (13%)
|
0
|
|
|
Streptomycin
|
2 (13%)
|
0
|
|
|
Para-aminosalicylic acid (PAS)
|
2 (13%)
|
0
|
|
|
Linezolid
|
2 (13%)
|
0
|
|
|
Clofazamine
|
1 (6%)
|
0
|
|
|
Adverse effects reported
|
13 (81%)
|
16 (33%)
|
< 0.001
|
|
Arthralgia
|
0
|
1 (2%)
|
1.0
|
|
Haematological abnormalities
|
2 (13%)
|
1 (2%)
|
0.13
|
|
Hearing impairment
|
4 (25%)
|
0
|
0.002
|
|
Hypothyroidism
|
1 (6%)
|
0
|
0.25
|
|
Injection site complications
|
3 (19%)
|
0
|
0.008
|
|
Liver dysfunction
|
3 (19%)
|
2 (4%)
|
0.13
|
|
Nausea/vomiting
|
11 (69%)
|
5 (10%)
|
< 0.001
|
|
Psychiatric problems
|
7 (44%)
|
0
|
< 0.001
|
|
Palpitations
|
1 (6%)
|
0
|
0.25
|
|
Paraesthesia
|
0
|
1 (2%)
|
1.0
|
|
Rash/itch
|
2 (13%)
|
10 (21%)
|
0.42
|
|
Renal dysfunction
|
1 (6%)
|
0
|
0.25
|
|
Tinnitus/vertigo
|
7 (44%)
|
0
|
< 0.001
|
|
Visual disturbance
|
1 (6%)
|
3 (6%)
|
0.68
|
|
Adverse effects requiring therapeutic change
|
5 (31%)
|
6 (13%)
|
0.02
|
|
Completion of therapy
|
|
|
|
|
Ongoing therapy
|
4 (25%)
|
3 (6%)
|
|
|
Transferred out before completion
|
3 (19%)
|
7 (15%)
|
0.72
|
|
Completed therapy
|
9 (56%)
|
38 (79%)
|
|
|
Mean total days of treatment (range)
|
597 (365–724)
|
229 (174–554)
|
|
|
Treatment outcome
|
|
|
|
|
Success*
|
9/12 (75%)
|
38/45 (84%)
|
0.72
|
|
Success in accessible cases†
|
9/9 (100%)
|
38/38 (100%)
|
|
|
Failed
|
0
|
0
|
|
|
Died
|
0
|
0
|
|
|
|
* Denominator excludes patients whose therapy was ongoing. † Denominator excludes patients whose therapy was ongoing and those who were transferred out before completion of therapy.
|