Haemoglobinopathies are inherited conditions caused by defects in globin chain synthesis. Worldwide, haemoglobinopathies are the most common genetic defect in humans — about 7% of the world’s population are carriers.1 In Australia, haemoglobinopathies are becoming more prevalent because of recent immigration of people from countries where these disorders are endemic.
Haemoglobinopathy disorders include α– and β-thalassaemia, sickle-cell disease and globin chain variants. Screening is complex and has limitations due to lack of evidence; ambiguities in the results of screening tests; health practitioners assessing patients’ risks with incomplete education in this evolving area; poor correlation between the genotype and phenotype in affected people leading to difficulty in predicting outcomes in an unborn child; definitive testing being expensive and not covered by Medicare; and generally lower health literacy in the target population.
Screening is predominantly initiated by general practitioners, obstetricians and midwives. Subsequently, haematologists, geneticists and laboratory scientists may become involved. In Australia, there is no national screening program and wide variation in testing practices. Many pregnant women are not being screened in a timely manner.2
In this article, we aim to provide a brief overview of the pathophysiology of haemoglobinopathies, a rationale for screening, and a description of the steps involved in screening and managing abnormal results. The term “haemoglobinopathy” is used broadly, rather than being restricted to variant haemoglobins.
Geographical distribution of haemoglobinopathies
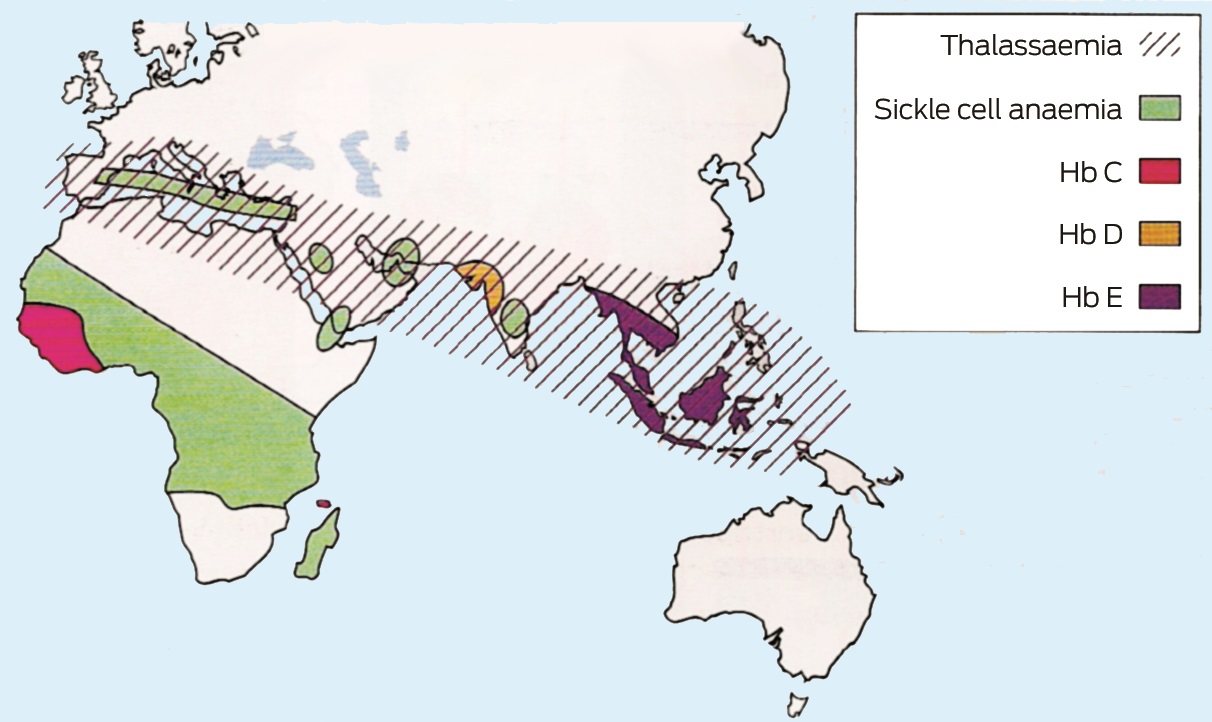
The incidence of α– and β-thalassaemia is high in Mediterranean countries, the Middle East, on the Indian subcontinent, in South-East Asia, and in parts of Africa. Severe forms of α-thalassaemia (αo) are more prevalent in South-East Asia. Sickle-cell anaemia has the highest incidence in tropical Africa. Haemoglobin E (HbE) is prevalent in South-East Asia and on the Indian subcontinent.1 Box 1 shows the geographical distribution of haemoglobinopathies.3 Migration from these regions to Australia is increasing.
Pathophysiology and clinical impact of haemoglobinopathies
Haemoglobin carries the oxygen in red blood cells. Structurally, it is a four globin chain tetramer (HbA, two α-globin chains and two β-globin chains). There are four α– and two β-globin genes. In adults, the major form of haemoglobin is HbA (α2β2) (about 95%); HbF (α2γ2) and HbA2 (α2δ2) are minor components (< 1% and 2–3% respectively). At birth, the major form of haemoglobin is HbF, which comprises 60–80% of total haemoglobin. At one year of age, adult levels of HbF and HbA are obtained.4 This makes the β-thalassaemia trait difficult to detect during infancy.
Decreased synthesis of globin chains results in thalassaemia (a quantitative defect). In α-thalassemia, reduced synthesis of α-globin chains leads to an excess of β-globin chains, and the situation is reversed in β-thalassaemia. The imbalance of globin chains causes haemolysis and impairs erythropoiesis. Conversely, structural changes in the globin chains are known as variant haemoglobins (qualitative defects). Box 2 summarises the clinical phenotypes and genotypes, and highlights the haemoglobinopathies that have significant clinical implications in the fetus, identifying those with transfusion dependency, shortened life expectancy or sickle-cell disease. The haemoglobinopathy trait is often asymptomatic and undetected, being clinically significant only if a woman is pregnant and her partner is also a carrier. Thalassaemia major and sickle-cell disease are managed in specialist centres.
The form of α-thalassaemia known as haemoglobin H disease produces a variety of symptoms ranging from none to transfusion dependency. Haemoglobin Barts leads to a non-viable fetus that dies in utero and significant maternal complications.5 Termination of pregnancy may be recommended, but there have been cases of intrauterine blood transfusions in Barts disease leading to a thalassaemia major phenotype after birth.6
β-Thalassaemia major renders an individual transfusion-dependent, which subsequently requires removal of excess iron (chelation). Without transfusions these patients die as children. Without iron chelation, death occurs in the second decade.7 Inadequate chelation leads to the complications of cardiomyopathy, cirrhosis, diabetes, hypothalamic hypogonadism, hypothyroidism and osteoporosis.8
The effects of sickle-cell disease are highly variable. The abnormal haemoglobin leads to “sickling” of the red blood cells, which causes occlusion of small blood vessels, leading to painful crises, acute chest syndrome (acute dyspnoea and hypoxia), splenic or bone infarcts and cerebrovascular thrombosis. Other problems include haemolytic crises and overwhelming pneumococcal infection from functional asplenia. This disease causes significant morbidity and the highest mortality rate is among 1–3-year-old children.7
Why and when should we screen?
Opportunities for screening include before conception, and the antenatal and neonatal periods.
Ideally, women should be screened before they conceive to allow those at risk to make informed decisions. Pre-conception screening also allows alternatives to termination, such as pre-implantation genetic diagnosis, gamete donation and adoption.9 Several countries with high carrier frequencies, such as Iran, Saudi Arabia, the Palestinian territories and Cyprus, have implemented mandatory pre-marital screening.6
Antenatal haemoglobinopathy screening aims to detect pregnancies at risk of carrying affected offspring, and should be performed early in pregnancy. In Australia, there is no national screening program and practice varies within states and local area health districts. In the United Kingdom, the National Health Service (NHS) Sickle-Cell and Thalassaemia Screening Programme aims to offer antenatal screening by 10 weeks’ gestation to enable prenatal diagnostic testing to be completed by 12 weeks’ gestation.10 Screening at the time that pregnancy is confirmed may require additional resources, but increases the number of pregnancies screened before 10 weeks’ gestation.11
A retrospective analysis of data of 462 patients of a large Australian antenatal service found that screening was performed in women at a median gestation of 15 weeks.2 A British study in a high prevalence area similarly found that 74% of women visited their GP early (at a mean gestation of 7.6 weeks); however, screening was delayed until a median gestation of 15.3 weeks, with only 4.4% being screened by 10 weeks.12 Evidence suggests that health professionals, rather than pregnant women, are responsible for the delay in screening. The barriers identified include the lack of time within the 10-minute consultation, delays in arranging blood tests, language problems and a lack of awareness among health professionals.13
Should any abnormality be identified, partner screening, genetic counselling and prenatal testing can be offered. Women in the earlier stages of pregnancy are more likely to accept prenatal diagnosis.14 By offering it sufficiently early in pregnancy, women can make informed, less time-pressured decisions, which might include termination of pregnancy. Termination can be offered for affected pregnancies up to 20 weeks’ gestation.7,15
In the UK, neonatal haemoglobinopathy screening occurs within the NHS Newborn Blood Spot Screening Programme16,17 aiming to identify infants with sickle-cell disease and β-thalassaemia major to improve outcomes through early treatment and care. New diagnoses lead to prenatal counselling before future pregnancies. In Australia, haemoglobinopathy screening is generally not part of the routine newborn screening program because of the lower prevalence of these disorders in this country. Effective maternal screening should abrogate the need for newborn screening.
The antenatal haemoglobinopathy screening process
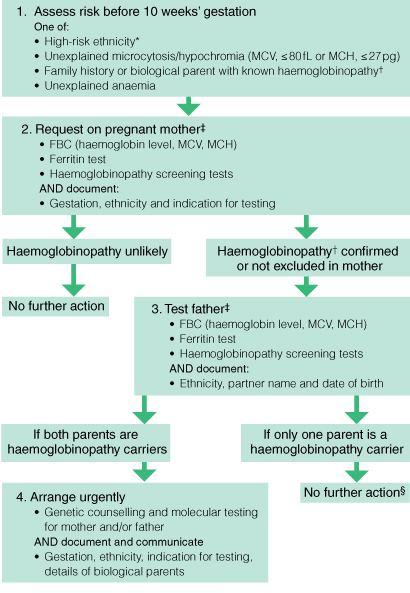
Box 3 shows a proposed algorithm for antenatal haemoglobinopathy screening.2 The diagnostic sequence for haemoglobinopathy is a multistep process. Local general practice guidelines state that all women should be screened with a full blood count and ferritin test during their first antenatal visit or during the pre-conception phase.18 Haemoglobinopathy screening should be performed on women:
-
with a mean corpuscular volume (MCV) of less than 80 fL, a mean corpuscular haemoglobin (MCH) mass of less than 27 pg, and a normal ferritin level (> 30 μg/L); or
-
with high risk features, such as belonging to a high risk ethnic group, having a family history of haemoglobinopathy, being the partner of a haemoglobinopathy carrier, or consanguinity with the partner.19
Screening tests for haemoglobinopathy are subsequently performed by various complementary methods. The use of molecular studies is determined by the maternal and paternal risk; however, there is significant variation in access and testing between health regions.
Full blood count and ferritin test
The haemoglobin level and MCH and MCV values can be used as a guide, but are not the sole indicators of the need for further screening. Most people with the α-thalassaemia trait (two gene deletions) have an MCH of less than 26 pg.19 Rarely, individuals may have the thalassaemia trait and a normal MCH, which could be due to a silent β-thalassaemia mutation, single α-gene deletion, or co-inheritance of both α– and β-thalassaemias.19 Structural variants, such as haemoglobin S (HbS; sickle-cell trait or disease) can be missed if a diagnosis is based on full blood count indices alone because people with HbS trait can have normal MCV and MCH values.
Although a low MCV is seen as a driver for further testing, MCH is more reliable because the MCV can increase artefactually because of red cell swelling if sample processing is delayed. Further, the MCV reference range differs between automated cell counters20 and it rises during pregnancy and in people with liver disease, and those with vitamin B12 or folate deficiency.
Ferritin testing helps with the interpretation of red cell indices, but iron deficiency cannot be used to exclude further screening, as thalassaemia can be found in iron-deficient women.19
Haemoglobinopathy screening tests
These include haemoglobin electrophoresis (HbEPG), high performance liquid chromatography (HPLC), and capillary electrophoresis (CE). They are described in Box 4.
In Australia, each laboratory performs at least two screening tests (most commonly HbEPG and HPLC) to identify and quantify HbA2, HbA, HbF and variant haemoglobins. HPLC and CE are the more accurate quantitative techniques.21
An elevated HbA2 level of 3.5–7.0% is diagnostic of β-thalassaemia trait.22 This is because reduced β-globin chain synthesis promotes the combining of excess α-globin chains with δ-globin chains. Rarely, silent β-thalassaemia can be missed because affected people have a normal or borderline HbA2 level.9 Iron deficiency has been thought to falsely lower the HbA2 level and obscure the diagnosis of β-thalassaemia. Data on this are conflicting, with one study concluding that there is a small but significant decrease in HbA2 level in iron-deficient patients, with no impact on making the diagnosis of the β-thalassaemia trait.20,23 Ideally, screening should be performed when patients are iron-replete. However, if time does not permit, screening tests can be performed and their results interpreted with caution. Further testing, either with genetic studies or partner screening, may be indicated in this setting.
Partner screening is offered to identified carriers or equivocal cases. Cost-effectiveness studies support screening of fathers sequentially rather than upfront testing,24 but this can lead to a time delay that may be critical in the antenatal setting.
Some laboratories offer combined couple risk assessment when results are available for both the mother and father, and if their relationship is clearly identified. This is clinically valuable to clinicians, but is time-consuming and assumes paternity.
Genetic diagnosis
A couple’s combined risk determines whether DNA testing for haemoglobinopathies is required. It is performed when α-thalassaemia cannot be excluded in both individuals, when there is uncertainty about the partner of a known β-thalassaemia carrier, and when both parents are affected and prenatal genetic diagnosis of the fetus is being pursued. The risk will change if there has been a change of partners between pregnancies. Genetic diagnosis is coupled with genetic counselling to discuss abnormal results.
Access to genetic testing for haemoglobinopathies and the costs and types of testing available vary. Genetic tests include multiplex ligation-dependent probe amplification, gap polymerase chain reaction and β-gene sequencing. Genetic testing does not have a Medicare Benefits Schedule item number, and the cost is borne by the patient, an institution, research grants, or the state government (Victoria only). It could be argued that α-thalassaemia gene testing should be publicly funded because thalassaemia carriers can have a low MCV and a normal HPLC result and ferritin level. Turnaround times are high (2–6 weeks), which reduces the clinical utility of antenatal testing if termination is being considered. There is no national registry for the results of genetic tests for haemoglobinopathies. In Victoria, only one laboratory performs the testing, which avoids duplicate testing in subsequent pregnancies, assuming identifiers are accurately correlated.
Prenatal diagnosis can be offered if there is a risk of the child being affected and if the parents’ genotypes are known or linkage analysis is possible. Fetal DNA obtained by chorionic villous sampling (CVS) at 11–13 weeks’ gestation or by amniocentesis from 15 weeks’ gestation onwards is tested. The risk of miscarriage is 0.5–1.0% for amniocentesis and 1–2% for CVS. In the future, it may be possible to perform prenatal diagnosis using free fetal DNA found in maternal plasma.25
Conclusion
Haemoglobinopathies are one of the most common genetic defects worldwide. In Australia, the number of carriers is increasing due to migration from countries with high carrier frequencies. Screening aims to reduce the burden of these disorders by identifying those at risk and managing their pregnancy choices, but there is currently no nationally coordinated screening program in this country.
To optimise clinical outcomes, screening should be performed before conception or early in pregnancy, and this requires awareness among patients and their doctors and carers, and short turnaround times for laboratories performing the tests.
Additionally, molecular testing should be more widely available. A national registry to record results is much needed to reduce duplicate testing and provide access to results. We envisage a future with cost-effective, accurate and accessible diagnostic testing. In the interim, prompt screening must be offered to at-risk pregnant women.
Box 1 –
Geographical distribution of haemoglobinopathies
Box 2 –
Haemoglobinopathies with significant clinical implications for the fetus
A. Summary of clinical phenotypes and genotypes of α– and β-thalassaemia
|
Disorder
|
Genotype
|
Anaemia
|
Clinical characteristics
|
|
|
α-Thalassaemia*
|
|
|
|
|
Silent
|
−α/αα
|
Absent
|
Nil; normal MCV
|
|
Minor (trait)
|
αα/− − or −α/−α
|
Mild
|
Asymptomatic; normal or reduced MCV
|
|
Haemoglobin H disease
|
α−/− −
|
Moderate
|
Variable; asymptomatic, microcytic anaemia, hepatosplenomegaly, may require transfusions
|
|
Major (haemoglobin Barts)†
|
− −/− −
|
Severe
|
Usually death in utero, hydrops fetalis syndrome
|
|
β-Thalassaemia‡
|
|
|
|
|
Minor (trait)
|
β/β0, β/β+
|
Mild
|
Asymptomatic; normal or reduced MCV
|
|
Intermedia
|
β+/β+, β0/β+
|
Moderate
|
Wide clinical variability; features range between those of thalassaemia trait and thalassaemia major
|
|
Major
|
β0/β0
|
Severe
|
Transfusion-dependent to prevent growth retardation, hepatosplenomegaly, haemolytic anaemia; iron overload from chronic transfusions, which reduced life expectancy before the use of iron chelation therapy
|
|
|
MCV = mean corpuscular volume. * Genotype nomenclature reflects the number of absent genes: α−/αα indicates one absent gene; α−/α− and αα/− − indicate two absent genes; α−/− − indicates three absent genes; and − −/− − indicates all genes absent. † α-Thalassaemia major is commonly referred to as haemoglobin Barts, although other mutation combinations can produce a similar phenotype. ‡ In genotype, β0 indicates absent β-globin chains and β+ indicates reduced synthesis of β-globin chains.
|
B. Summary of clinical phenotypes and genotypes of common structural variants of thalassaemia
|
Disorder
|
Genotype
|
Anaemia
|
Clinical characteristics
|
|
|
Haemoglobin S (HbS)
|
|
|
|
|
Sickle-cell trait
|
AS
|
Absent to mild
|
Mostly asymptomatic; normal MCV
|
|
Sickle-cell disease
|
SS, SC, S/βthal, SD, S/O-Arab
|
Mild to moderate
|
Vaso-occlusive phenomena and haemolysis affecting brain, chest, kidneys, bones and spleen
|
|
Haemoglobin E (HbE)
|
|
|
|
|
Trait
|
AE
|
Normal
|
May have mild microcytosis; no anaemia
|
|
Homozygous E
|
EE
|
Mild
|
Mild anaemia and microcytosis
|
|
Compound heterozygous
|
E/βthal
|
Moderate to severe
|
Variable phenotype, thalassaemia intermedia or thalassaemia major
|
|
Haemoglobin Constant Spring (HbCS)*
|
|
|
|
|
Heterozygous
|
αα/αCSα
|
Mild
|
Asymptomatic
|
|
Homozygous
|
αCSα/αCSα
|
Moderate
|
Splenomegaly, moderate haemolytic anaemia
|
|
Compound heterozygous (in conjunction with α0 thalassaemia)
|
− −/αCSα
|
Moderate to severe
|
Wide clinical variability; often more severe than deletional haemoglobin H disease, with possible transfusion dependence
|
|
|
MCV = mean corpuscular volume. * First isolated in the Constant Spring district of Jamaica and characterised by a variant of the α-globin chain (designated αCS), which is abnormally long.
|
Box 3 –
A selective screening algorithm for antenatal haemoglobinopathy (adapted from Lavee et al2)
Box 4 –
Specialised laboratory methods for haemoglobinopathy screening
|
Test
|
Description
|
|
|
Haemoglobin electrophoresis (HbEPG)
|
- Separation of haemoglobins with electrophoresis at pH 8.4 (alkaline) and pH 6.2 (acid)
- Identifies variant haemoglobins, but is not useful for quantitative purposes
- Does not detect α-thalassaemia trait
- Cheap, laborious, operator-dependent
|
|
High performance liquid chromatography (HPLC)
|
- Separates haemoglobins based on adsorption and ion exchange when a blood sample is in contact with a mobile liquid phase and a solid stationary phase
- Identifies and quantifies HbS, HbA2, HbF and variant haemoglobins
- A raised HbA2 level (3.5–7.0%) indicates the presence of β-thalassaemia trait
- The HbA2 level may not be accurately quantified in the presence of HbS
- Does not separate HbA2 from HbE
- Does not detect α-thalassaemia trait
- Automated, quick, small sample volume required (5 μL)
|
|
Capillary electrophoresis (CE)
|
- Haemoglobin variants are separated by electro-osmotic flow with negatively charged silica capillaries and high voltage
- Can be used as a complementary method to HPLC
- Quantitative method
- Separates HbE from HbA2
- Does not detect α-thalassaemia trait
|
|
HbH inclusions
|
- Red cells are stained to look for HbH inclusions (tetramers of β-globin chains)
- Diagnostic for α-thalassaemia trait, HbH disease, α-thalassaemia and mental retardation (ATRX syndrome) or acquired HbH disease
- HbH bodies are much easier to find in αo heterozygosity
- α-thalassaemia trait is not excluded with a negative test result
- Operator-dependent
|
|
Sickle solubility test
|
- Sodium dithionite/metabisulfite reduces the amount of oxygen present in the blood sample, causing abnormal red cells to sickle
- Gives a positive result in HbS trait and HbS disease
- Not routinely performed; has been replaced by HPLC
|
|
|
ATRX = α-thalassemia mental retardation; Hb = haemoglobin.
|