Precision genomic medicine will have a transformative impact on personal health and wellbeing, health economics and national productivity
In recent years, there has been an extraordinary leap in knowledge of the human genome and its role in health and disease. A decade ago, researchers were tentatively exploring the first reference human genome sequences, which cost over $1 billion to produce.1,2 Now, thousands of genomes from a cross-section of ethnic backgrounds have been sequenced. This explosion of activity has been enabled by unprecedented advances in sequencing technologies that can now sequence a person’s entire genome — more than 6000 million bases — in days, at a cost of US$1000,3 with costs expected to fall further in coming years.
Making sense of genomic data requires computational technologies and databases to evolve in parallel with sequencing technologies. Advances in both technologies enable an ever-increasing capacity for accurate diagnosis of existing disease, and development of effective and targeted treatment strategies. They also offer opportunities to assess predisposition to disease, potentially prompting more focused clinical monitoring and lifestyle changes.
Although our knowledge of the human genome is currently far from complete, accumulating examples demonstrate that even our limited genomic understanding can be powerful in the clinic. Currently, genome sequencing is having the greatest impact in stratifying cancer, characterising genetic disease, and providing information about an individual’s likely response to treatment.
Cancer: stratifying tumours for treatment
Genomic medicine has already shown benefit in refining diagnoses and guiding therapeutic approaches for cancer.4 Since the late 1990s, the clinician’s cancer “toolkit” of surgery, radiation and chemotherapy has been increasingly supplemented by therapies that target specific molecular pathways in cancer growth and development.5 Genomic information can now assist clinicians in deciding treatment strategies by classifying a tumour according to its mutations and corresponding drug sensitivities. In some cases, patients have been spared costly and complex procedures, such as bone marrow transplants, based on a molecular diagnosis.6 In other examples, a patient’s cancer development has been stabilised — for a time at least — by targeting specific molecules or pathways in the tumour cells.7 More controversially, clinicians are starting to use genomic information to refine a cancer diagnosis and prognosis and alter an individual’s quality of life, even when therapies are not currently available.8
Genome-wide sequencing is now also being applied to the analysis of circulating DNA in the plasma of cancer patients, as well as in individuals with other diseases.9 This technology enables non-invasive tumour detection and monitoring responses to therapy that promise to significantly improve patient management.
Drug prescription and development
Cancer treatment is also set to benefit from genomic information to predict how an individual will respond to drugs (known as pharmacogenomics) and inform prescription of the appropriate drug or dosage. Pharmacogenomic applications extend into many areas of clinical practice; for example, in the prescribing of drugs such as antidepressants, analgesics and anticoagulants.10
In the longer term, genomic information is expected to dramatically change the testing and use of pharmaceuticals through disease stratification.11 New research into molecular pathways underlying health and disease will continue to inform rational drug development and design. In parallel, researchers are using genomic data to suggest new therapeutic applications for existing drugs (repositioning) — with significant cost savings — and to better select individuals for clinical trials to find uses for drugs that failed earlier trials involving broader cohorts (rescue and repurposing).12 Better targeting of existing drug use avoids wasteful and risky therapy and can provide savings for the health care system and the economy at large.
Diagnosing and characterising genetic disease
With the exception of a few pharmacogenomic tests and cancer treatments, clinical genetic testing in Australia is at present limited to diagnosing hereditary and rare monogenic diseases. Every baby born in Australia is offered screening for about 30 genetic conditions in the Guthrie test, and more than 300 tests for genetic disorders are available through the health care system.13 However, clinicians worldwide are currently embracing far more comprehensive genome sequencing to search for variants implicated in undiagnosed genetic diseases14,15 and are using this information to guide treatment.16 As the field matures and our understanding of the affected pathways improves, there will be many more genomic loci implicated in rare disease, with improved treatment prospects. Further, genome sequencing is increasingly being used to assess genetic contributions to complex diseases, where multiple gene variants may be involved in disease development and progression.
Rather than sequencing the entire genome, rapid sequencing of the protein-coding portion (the exome, about 1.5% of the total) can offer comparatively cost-effective analysis of many genetic disorders. Although less effective in detecting chromosomal duplications, deletions and rearrangements, exome sequencing identifies most clinically relevant nucleotide variants in a single analysis, serving both as a diagnostic tool and a method to discover new genes and mutations that underlie these classes of disease. However, as the function of intergenic and intronic regions in disease-regulatory processes is further elaborated and variants in these regions are shown to have diagnostic utility, whole genome sequencing is likely to provide a clinically more robust, accurate and useful test that will replace exome sequencing as sequencing costs decrease.
From disease diagnosis to personalised genetic health
With genome sequencing becoming more accessible, it is becoming more common for individuals to seek personal genomic information through direct-to-consumer genetic testing services.17 However, these tests are not accredited for clinical diagnosis, and individuals will gain most benefit when their genomic sequence becomes integrated into their health care such that clinicians and other health professionals can provide considered advice and the portal to treatment options.
Inevitably, personal genome sequences, likely obtained at birth, will become an integral part of a patient’s electronic health record (EHR), where this information will be integrated with other clinical and environmental data and interrogated throughout the individual’s lifetime. Clinicians and patients may then query the sequence to accurately prescribe treatments, determine disease susceptibilities and identify drug sensitivities, and to determine a course of action to monitor, manage, ameliorate risk of or prevent the disease.
The idea of sequencing newborns remains controversial, because of questions about consent, potential stigmatisation and the value of the information to the individual.18 There are also practical questions about data storage and access, and the question of which results are returned when and to whom. Valuable insights are expected from projects recently funded in the United States exploring the use of genomic sequencing in paediatric medicine and the ethical issues that arise.19
Integrating genomic and clinical information
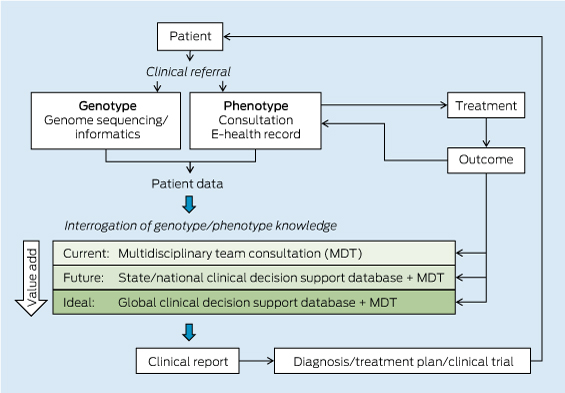
The pathway from patient DNA sequencing to a clinical treatment plan relies on the integration of the individual’s genomic information with knowledge databases that contain known genotype–phenotype correlations and genomic and clinical associations from large populations of individuals (Box 1).
Clinical decision making currently relies on the knowledge of individual practitioners and genetic counsellors. These practitioners work as a multidisciplinary team to assess the genomic variants identified through genomic sequencing and arrive at a treatment plan. Purpose-built, well curated and continuously updated evidence-based databases of human genotype–phenotype associations are urgently needed, with computational tools to interrogate the ever-increasing information in an automated way.
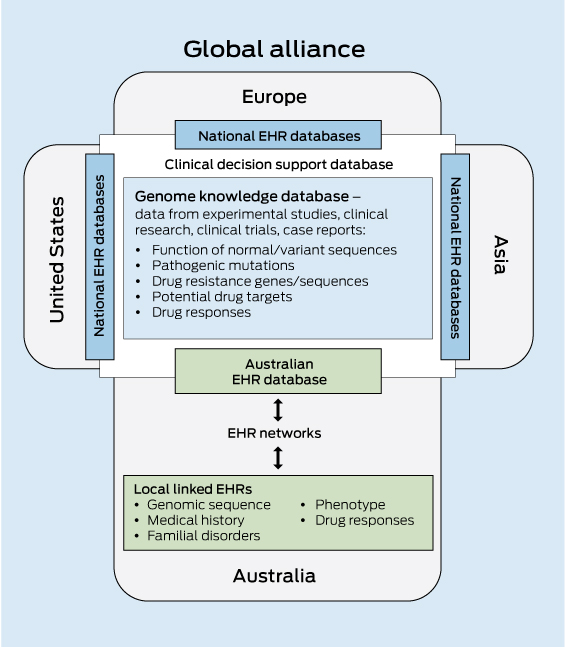
The inherent computational challenges in integrating genomic and clinical data necessitate a significant investment in bioinformatics capability. Given the considerable overheads of both storage and computational power, Australia will most effectively be served by a centralised genome knowledge repository linked to global repositories. Infrastructure support will also be critical for the development of e-systems and software interfaces. These must enable clinicians and health care providers to interrogate a patient’s genomic sequence against the clinical decision support databases to obtain an informative clinical report (Box 2).
Storing and sharing population data
The speed with which personalised genomic medicine becomes a reality for most of the population will be strongly influenced by the sharing of clinical information — in this case, an individual’s genomic sequence together with their medical record — as part of large population datasets that are accessible to clinicians and translational researchers. By aggregating and analysing large datasets, it will be possible to uncover patterns and relationships that would not otherwise be evident. The enormous value of data sharing in the acceleration of progress in genomic medicine is now recognised. A major role of the international Human Variome Project (http://www.humanvariomeproject.org) is to ensure that genetic variation information generated during routine diagnostic and predictive testing is collected and shared in the course of routine clinical practice. In addition, a consortium of 125 institutions in 40 countries have formed the Global Alliance for Genomics and Health to develop a framework for data sharing, including the technical standards required for storage and sharing of genomic and clinical data, and for the management of privacy, informed consent and security.20
Adding genomic information and linking large population datasets to a personally controlled EHR system would add significant value to the health care system through the acceleration of personalised genomic medicine. Typically, only a small subset of the genetic information yielded in a genomic test is used for clinical diagnosis, so this could become a potential treasure trove of information for translational researchers to further understand human variation and disease. Data mining of a national EHR database will allow evidence-based forward planning of health care needs and allocation of resources by government health departments. As a national resource, it is vital that such a database is access-controlled and that appropriate consent processes are in place.
Economic benefits of genomic medicine
Genomic medicine will transform health care and the national economy, especially in a population whose average lifespan is increasing. Personal economic benefits accrue from genomically informed restoration of health and consequent earning capacity. Higher precision in risk identification reduces health costs for an individual and the health care system by avoiding adverse reactions and unnecessary treatments.
Genomic medicine has the potential to make genetic diagnosis of disease a more efficient and cost-effective process, by reducing genetic testing to a single analysis, which then informs individuals throughout life. Although individuals will vary in their response to genomic information,21 personal identification of risk could be expected to result in uptake of more effective monitoring and preventive actions.
Genomic information and its application to technical developments, medical research and health care will also have a major impact on the national economy, not only by reducing productivity losses and decreasing costs of treating disease, but also by creating new medical information industries.
The rapid increase in the age of populations, accelerating costs of health care, and the growing burden of chronic disease present major challenges to health systems worldwide. The UK government has recognised this imperative, recently announcing its investment in a project, to be run by Genomics England, to sequence the genome of 100 000 patients over 5 years and to introduce genomic technology into its mainstream health system.22
Opportunities and challenges for the future
For clinical genomics to provide full benefit for the Australian community, major imperatives are to:
- build the infrastructure for EHR databases that integrate patients’ genomic and medical information for clinical and research applications, with appropriate mechanisms for patient consent, protection of privacy and data security;
- establish national and international knowledge-sharing platforms using a standardised approach for recording, sharing and interrogating fully integrated clinical and genomic databases to provide clinically useful reports; and
- develop well designed and integrated public and professional education efforts nationwide to engage clinicians, the health workforce and the community in fully realising the medical potential of genomics.23
Genomic analysis provides opportunities for new approaches to therapeutic development, health care delivery and population health management. The medical and scientific communities around the world are just starting to seize the transformative opportunities that personalised, precision genomic medicine offers. With further investment in the infrastructure required to acquire and share clinical and genomic data, Australia will be positioned as one of the key leaders and major beneficiaries of the implementation of genomic medicine.
1 The patient pathway in personalised genomic medicine
2 Components of a clinical decision support database for genomic medicine