Acronyms
6MWD 6-minute walk distance
BMPR2 bone morphogenetic protein receptor type 2
cGMP cyclic guanosine monophosphate
CTEPH chronic thromboembolic pulmonary hypertension
ERA endothelin receptor antagonist
iPAH idiopathic pulmonary arterial hypertension
NYHA New York Heart Association
PAH pulmonary arterial hypertension
PBS Pharmaceutical Benefits Scheme
PDE5 phosphodiesterase type 5
PEA pulmonary endarterectomy
PH pulmonary hypertension
PVR pulmonary vascular resistance
RCT randomised controlled trial
TCD time to clinical deterioration
TGF-β transforming growth factor beta
VQ ventilation–perfusion
In contrast to the systemic circulation, the pulmonary circulation is a low-pressure circuit, which normally operates with a mean pressure below 20 mmHg. Pulmonary hypertension (PH) is defined haemodynamically by an elevated mean pulmonary artery pressure of ≥ 25 mmHg at rest. The presence of PH, irrespective of the cause, is associated with poor prognosis,1 often due to right heart failure in the face of persistently elevated afterload. There are multiple causes of PH, which has been classified into five groups according to the initiating cause and whether the predominant part of the pulmonary circulation affected is pre-capillary or post-capillary (Box 1).2
Pulmonary arterial hypertension (PAH; Group 1 PH) is caused by vascular proliferation and obstruction predominantly affecting the arterial side of the pulmonary circulation. PAH is defined as PH with a normal left atrial pressure (pulmonary artery wedge pressure ≤ 15 mmHg) and an elevated pulmonary vascular resistance (PVR) > 3 Wood units in the absence of other causes of pre-capillary PH. The most common forms are idiopathic PAH (iPAH), familial PAH and PAH secondary to connective tissue disease, most commonly systemic sclerosis. PAH is rare, with a prevalence between 16 and 26 cases per million people, but iPAH has a dismal prognosis if left untreated, with a median survival of only 2.8 years.3 Chronic thromboembolic pulmonary hypertension (CTEPH; Group 4 PH) occurs due to incomplete resolution of pulmonary embolism, with subsequent remodelling of pulmonary arteries. Most such episodes of pulmonary embolism are clinically overt, but can be clinically silent.4
When PAH was last reviewed in the MJA,5 there were very few treatment options available. Over the past 10 years, many oral and additional intravenous therapies that have improved the prognosis for those with this condition have become available in Australia. For this review, we searched PubMed for original and review articles from 1984 to 2016, as well as specialist society guidelines, to formulate an evidence-based overview of PAH treatment as applied to clinical practice. We focus on Group 1 and Group 4 PH, for which there is good evidence for pharmacological treatment. While we acknowledge that Group 2 PH is the most common type in our community,1 there is no convincing evidence of benefit for treatment directed at Group 2 or Group 3 PH.
Old and new pathophysiological concepts in pulmonary arterial hypertension
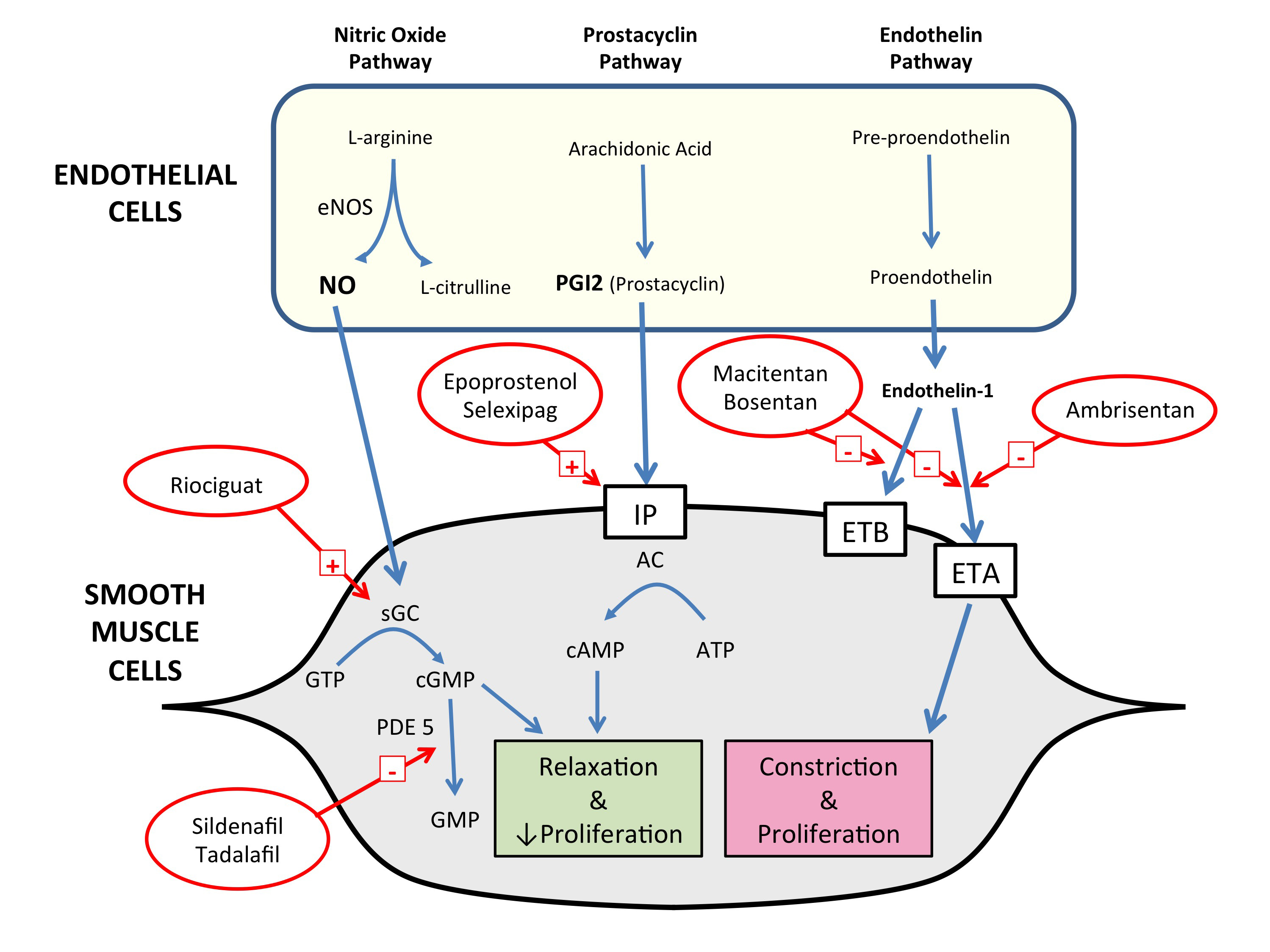
PAH is a disease of endothelial dysfunction and abnormal remodelling of the pulmonary vessels, leading to luminal narrowing, loss of normal vasodilator responses and obliteration of pulmonary arterioles, all resulting in elevated PVR. There is migration and proliferation of vascular smooth muscle cells, appearance of myofibroblasts and increases in extracellular matrix, leading to intimal hyperplasia, medial hypertrophy and adventitial fibrosis. Inflammatory infiltrates are a histological feature of PAH.6 It is thought that in situ thrombosis may also lead to vessel loss.7 It has long been recognised that three major endothelial signalling systems within the vessel walls are dysfunctional in PAH, and these have been the major targets for drug therapies (Box 2). Increased activity of the endothelin system promotes vasoconstriction and vascular smooth muscle cell proliferation.8 A further mechanism important in iPAH is reduction in the synthesis and availability of nitric oxide, which has potent pulmonary vasodilator and antiproliferative effects.9 The endothelial cells in iPAH demonstrate a reduction in prostaglandin I2, another vasodilator with antiproliferative properties.10 While the disease primarily affects blood vessels, it generally results in death due to right heart failure.11
Recently, there has been interest in the factors responsible for initiating and driving the process that results in PAH. The most common cause of familial PAH is mutations in the bone morphogenetic protein receptor type 2 (BMPR2) gene, a member of the transforming growth factor beta (TGF-β) signalling family and thought to be responsible in 80% of cases, although mutations associated with PAH have been described in other genes in the TGF-β superfamily.12 However, there have been many specific BMPR2 mutations identified, with the most common accounting for only 8% of familial PAH, making genetic screening for this disease impractical. Not all carriers of the abnormal genes develop PAH, suggesting additional influences are necessary for phenotypic expression, possibly reflecting a “multiple hit” process, similar to that seen with many cancers.13 It has been shown that BMPR2 is downregulated in other forms of PAH, suggesting that this may also be important in non-inherited forms of PAH. Dysregulated immunity and inflammation may also be important in the development of PAH.14 Patients with PAH have been observed to have elevated levels of serotonin. Disruption of the normal serotonin transporter affects the development of PH in animal models, signifying that the interaction between serotonin and vascular smooth muscle cells may be important in the development of PAH.6,14
Chronic thromboembolic pulmonary hypertension
In a small proportion of patients who experience an acute pulmonary embolism, the thrombus will fail to resolve afterwards. This was initially thought to be true for about 0.5% of patients with pulmonary embolism, but a landmark report suggested almost 4% of patients will develop evidence of CTEPH in the 2 years after a pulmonary embolism.15 Patients with CTEPH may typically present with only partial recovery from a pulmonary embolism, followed by a (“honeymoon”) period of stability (which may last months or even years), then subsequent decline. At least 25% of patients have no history of an acute embolus, presenting with progressive breathlessness.4 Although increased propensity to thrombosis seems to predispose to CTEPH (32% of patients in a large international registry had a thrombophilic disorder),4 the underlying abnormality appears to be related to abnormal thrombin or abnormal thrombinolysis — a clot forms in the pulmonary artery but is incompletely broken down, resulting in a complex obstructing lesion in the artery.
Survival at 3 years without surgery is 70%, but this rises to 89% if surgery is performed.16 Although no randomised controlled trial (RCT) has been conducted, pulmonary endarterectomy (PEA), where feasible, is regarded as the treatment of choice, and initial investigations should focus on suitability for PEA. The presence of PH may be evident on echocardiography, but confirmation requires right heart catheterisation.17 Persistent segmental or larger unmatched perfusion defects seen on a nuclear ventilation–perfusion (VQ) scan are highly suggestive of operable CTEPH (a negative VQ scan virtually excludes operable CTEPH, except where the pulmonary artery trunk or both the left and right pulmonary arteries have high grade obstruction).17 A negative computed tomography pulmonary angiography scan does not exclude operable CTEPH and, as such, is not the best screening modality. Pulmonary angiography is currently the gold standard for assessing the extent and surgical accessibility of the obstructing lesions.18
PEA performed in experienced centres carries a surgical mortality of 2–5%.19,20 Most patients return to near normal functional capacity (New York Heart Association [NYHA] Class I or II) after surgery.21 PEA involves extensive endarterectomy from the main pulmonary artery down to segmental branches. It is performed under hypothermic circulatory arrest (18°C) in a few expert centres in Australia.
In patients with persistent PH after PEA, or in those unsuitable for surgery, selective pulmonary vasodilators may be useful. For most selective vasodilators, case series have shown at least haemodynamic improvements. In a 16-week RCT, riociguat (a novel soluble guanylate cyclase stimulator) resulted in haemodynamic improvements, as well as a significant increase in 6-minute walk distance (6MWD) of 46 m.22 The 6MWD is a simple test performed under standardised conditions of the distance walked over flat ground in 6 minutes; it is often used in PH trials. Concern remains that patients who are suitable for surgery should not be initially offered vasodilator therapy, as both the drug and the delay to PEA may lead to an inferior outcome.17 Pulmonary artery balloon angioplasty may offer a viable alternative to PEA in some circumstances, where several discrete, more distal lesions are inaccessible surgically. However, the role of balloon angioplasty as a substitute for PEA is currently unclear, especially in older patients who are less well suited to, or accepting of, major surgery. More systematic study of this matter is warranted.23
Current drugs for pulmonary arterial hypertension
The existing treatments for iPAH target signal pathways proven to be involved in the disease’s pathophysiology (Box 2). Ultimately, normal life expectancy and normal quality of life are the objectives of therapy. Realistically, moving patients to a low risk of mortality with, at worst, mild functional limitation (NYHA Class I–II) is the present target of therapy.24
Patients whose PH may be Group 1 or Group 4 in aetiology should be referred to a specialised PH centre for diagnosis and treatment. Patient education and use of expert centre support are essential for optimal outcomes and minimal side effects using currently available drug treatments (Box 3).
Calcium channel blockers
There is a small group of patients with iPAH (6%) who appear to respond well to high doses of dihydropyridine calcium channel blockers and who may have durable benefit from this therapy.25 These patients are identified using a vasodilator challenge at the time of right heart catheterisation. Patients with no acute vasodilator response should not be treated with high dose calcium channel blockers.
Endothelin receptor antagonists
The oral endothelin receptor antagonists (ERAs) bosentan and ambrisentan (and the newer ERA macitentan) inhibit the signal pathway by blocking endothelin receptors. Monotherapy with bosentan and ambrisentan has produced improvements in 6MWD, PVR and time to clinical deterioration.26,27
Phosphodiesterase type 5 inhibitors
Phosphodiesterase type 5 (PDE5) breaks down cyclic guanosine monophosphate (cGMP) and therefore decreases the vasodilator effects of nitric oxide. The oral PDE5 inhibitors sildenafil and tadalafil decrease cGMP degradation, which increases the activity of the nitric oxide system and clinically improves 6MWD in patients with iPAH.28,29 Tadalafil also increases time to clinical deterioration compared with placebo.29
Prostanoids
The two major prostanoids used in Australia are epoprostenol and iloprost. Epoprostenol is the most effective single agent for treatment of PAH, but it requires delivery of a continuous intravenous infusion via a central venous catheter. It has been shown to improve 6MWD, haemodynamics, quality of life and survival.30 Recently, a more thermostable formulation of epoprostenol has become available, which may be more convenient to use as it eliminates the need for a cold pack to maintain the drug’s temperature at the required level. Inhaled iloprost improves functional class, 6MWD and time to clinical deterioration,31 but requires up to nine inhalations per day.
Anticoagulants
Early pathological studies showed thrombosis within small lung vessels in patients with PAH,32 leading to treatment with anticoagulants becoming part of standard therapy. Anticoagulation in patients with iPAH may be of benefit, with some observational series indicating improvements in survival, but uncertainty persists due to the low level of evidence.7,25,33 Use of anticoagulation in patients with PAH and scleroderma is more controversial.34 Recent guidelines for the treatment of iPAH state that oral anticoagulation with warfarin may be considered in treatment algorithms.35
New drugs for pulmonary arterial hypertension
While calcium channel blockers, intravenous epoprostenol, the oral ERAs bosentan and ambrisentan, and PDE5 inhibitors sildenafil and tadalafil are well established in the treatment of iPAH in Australia, new drugs have recently been introduced or will soon be available (Box 3). Direct comparison with currently available treatments is difficult, as most current treatments were tested in relatively small trials of short duration, using surrogate endpoints based largely on haemodynamic or functional measures. In contrast, trials of newer agents have been larger and in many cases have used mortality and morbidity endpoints, such as time to clinical deterioration, to demonstrate efficacy. Direct comparison is also difficult due to the widespread use of background PAH therapies in recent studies.
Macitentan
Macitentan is a recently introduced non-selective ERA developed with the aims of improving effectiveness and reducing the side effects, toxicities and drug interactions associated with earlier ERAs. Macitentan has been assessed in the SERAPHIN trial, the largest RCT of ERAs, in which two macitentan doses (3 mg and 10 mg) were compared with placebo in 742 patients with Group 1 PH over a median treatment period of 115 weeks.36 Occurrence of the primary outcome (first occurrence of a composite of death, lung transplantation, atrial septostomy, initiation of intravenous or subcutaneous prostanoids, or worsening of PAH) was reduced by 30% (P = 0.01) with 3 mg and by 45% (P < 0.001) with 10 mg of macitentan. This was mostly driven by a reduction in occurrence of worsening of PAH. Worsening of PAH was defined as a decrease in 6MWD of at least 15%, worsening of PAH symptoms and the need for additional PAH treatment. Reductions in hospitalisation were also seen with macitentan treatment. About two-thirds of patients in this study were already taking other PAH treatments, predominantly PDE5 inhibitors, with the remaining third taking the study drug as monotherapy. Improved outcomes with macitentan were seen in both the treatment-naive patients and those using combination therapy.36
Riociguat
The effectiveness of PDE5 inhibitors may be limited because PAH is a state of nitric oxide deficiency, and inhibiting breakdown of the already reduced levels of cGMP may not be the most effective strategy. Riociguat acts directly to stimulate soluble guanylate cyclase and thus increase cGMP independent of nitric oxide levels.37 Riociguat has been tested in patients with Group 1 PH in the PATENT-1 trial.38 This study compared two doses of riociguat with placebo in 443 patients, of whom half were treatment naive. Most of those taking the active drug were treated with 2.5 mg three times a day (254 patients). Riociguat resulted in a placebo-corrected increase in 6MWD of 36 m after 12 weeks of treatment (P < 0.001). In addition, improvements were seen in PVR, time to clinical deterioration, functional class, N-terminal pro-brain natriuretic peptide levels and quality of life with riociguat treatment.38
Selexipag
Although intravenous epoprostenol is a highly effective treatment for PAH, the logistics and infective complications associated with chronic intravenous administration limit its use.30 Other intravenous, oral and subcutaneous prostanoids have been limited by difficulties in their administration or intolerable side effects.31,39 More recently, a non-prostanoid prostacyclin receptor agonist, selexipag, has been studied in patients with PAH. The 1152 patients with Group 1 PH in the event-driven GRIPHON trial were randomly allocated to receive selexipag or placebo and followed for a median of 70.7 and 63.7 weeks, respectively.40 About 80% were already using specific PAH therapies, and the remaining 20% were treatment naive. Selexipag was uptitrated to 1600 μg twice daily or the highest tolerated dose. Selexipag treatment was associated with a 40% lower risk of the primary composite endpoint of death, hospitalisation for PAH, disease progression, initiation of parenteral prostanoid or long term oxygen therapy, or lung transplantation. The magnitude of benefit did not differ across different achieved doses, suggesting that the maximum tolerated dose is the correct dose for the patient. Several side effects, including jaw pain, nausea, diarrhoea, vomiting and myalgia, were more common in those treated with selexipag.
Combination therapy
In diseases such as cancer, human immunodeficiency virus, systemic hypertension and heart failure, treatment with combinations of drugs has proven to be more effective than treatment with a single agent. There is now evidence that combining specific therapies for PAH is more effective than monotherapy. Combination therapy may be used either sequentially or as initial therapy at the time of diagnosis. The strongest evidence for initial combination therapy comes from the AMBITION trial, which showed that an initial combination of ambrisentan and tadalafil was superior to either agent alone, with a 50% reduction in the occurrence of a primary endpoint event (first event of clinical failure) for those taking combination therapy compared with the monotherapy group.41 There were also beneficial effects on secondary endpoints.
In recent trials of new therapies, the rate of background PAH therapy has been significant, with about 80% of patients in the GRIPHON trial of selexipag,40 50% of patients in the PATENT-1 trial of riociguat,42 and 64% of patients in the SERAPHIN trial of macitentan36 having the trial drug added to background PAH therapy. Benefit has been observed in both those taking background therapy and treatment-naive patients, suggesting new drugs should be added to existing treatments, rather than substituted for them; a position that is supported by current international guidelines and consensus statements.35,43 A small pilot study in France showed excellent outcomes with initial triple combination therapy, including an intravenous prostenoid.44 A large study of initial combination double versus triple therapy (TRITON), comparing macitentan and sildenafil with selexipag or placebo, will commence recruitment in the next few months and we hope will help define the role for more aggressive initial oral therapies.
Access to treatment in Australia
There have been no head-to-head clinical trials of sufficient size or quality to make any scientifically based judgement on which of the available drugs can be considered first-line therapy for PAH, and local practice around the world is often based on cost. It is also not known how many patients in Australia are using each type of therapy, as the databases that exist likely do not reflect the whole country.
Two issues affect patient access to specific PAH therapies in Australia. While recent trial data indicate that combination therapy is superior to monotherapy, currently only treatment with one agent at a time is funded through the Pharmaceutical Benefits Scheme (PBS). However, based on our experience, we are aware that a significant number of Australian patients are using more than one PAH treatment, some of which is self-funded, some funded through hospitals and some sourced through compassionate access schemes. There are no systematic data on the use of combination therapy by patients.
Recent changes to the PBS have simplified the ongoing treatment of patients with PAH, with a reduction in administrative and repeat testing burden. Patients who have commenced PAH treatment or who have changed drugs must demonstrate disease stability (as described in the PBS explanatory notes) or improvement using 6MWD and echocardiography and/or right heart catheterisation after 6 months of treatment. Ongoing treatment at 12 months and beyond will no longer require demonstration of stability. Repeat assessment should, however, continue to be performed on clinical grounds and in accordance with practice guidelines.35
Future treatments
Present therapies rarely induce remission and never cure, to use a cancer analogy, and overall 3-year survival with their use is about 75%.36 Alternative treatment pathways identified through basic research are being evaluated. Initial enthusiasm for the tyrosine kinase inhibitor imatinib has been dampened by the high rate of side effects in a clinical trial setting.45 Several other compounds are in pre-clinical or early clinical evaluation.46
For patients whose PAH is not well controlled using available therapies, transplantation (double lung or heart–lung) remains an option. With improvements in organ availability in Australia, this is both feasible and likely; however, chronic lung allograft dysfunction still limits median survival to about 8 years.47 Developments in regenerative medicine may allow repopulation of a donated lung (rendered acellular) with autologous stem cells,48 an exciting approach currently in pre-clinical development.
The recognition that up to 30% of patients with apparently idiopathic PAH have an identified gene defect raises the prospect of gene therapy.49 Mutations of the BMPR2 gene are the commonest, and correcting this defect with gene therapy has been suggested — using an adenovirus, a vector can transfect the relevant cell.50 However, this leads to an immunological response, which means this approach will be unsuitable for recurrent treatments. Endothelial progenitor cells may help repair the vascular endothelium, and there are data from human studies on this approach.51 More recently, the use of endothelial progenitor cells as vectors for gene therapy has been proposed, with a human study recently attempting to introduce an inducible nitric oxide synthase (iNOS) gene into the pulmonary vascular endothelium.52
Conclusion
The past 10 years have seen the introduction into routine clinical practice of new oral and intravenous therapies for PAH, based on the endothelin, nitric oxide and prostacyclin pathways, leading to improvements in functional capacity and survival. Emerging evidence supports more widespread use of combination therapy, as both initial and sequential treatment, although there are significant barriers to this use in Australia. CTEPH remains an important cause of PH to identify because of its potential for successful treatment using surgery, medication and possibly angioplasty. There is enthusiasm for exploring new pathways that are important in PAH to identify new agents that may address more basic elements of pathophysiology and arrest or reverse the process of pulmonary blood vessel obstruction and obliteration.
Box 1 –
Simplified World Health Organization classification of pulmonary hypertension*
- 1. Pulmonary arterial hypertension (PAH)
- 1.1 Idiopathic PAH
- 1.2 Heritable PAH (BMPR2, ALK1, ENG, CAV1 and other mutations)
- 1.3 Drug- and toxin-induced pulmonary hypertension
- 1.4 PAH associated with:
- 1.4.1 Connective tissue disease
- 1.4.2 HIV infection
- 1.4.3 Portal hypertension
- 1.4.4 Congenital heart disease
- 1′. Pulmonary veno-occlusive disease (PVOD) and/or pulmonary capillary haemangiomatosis (PCH)†
- 2. Pulmonary hypertension due to left heart disease
- 2.1 Left ventricular systolic dysfunction
- 2.2 Left ventricular diastolic dysfunction
- 2.3 Valvular disease
- 2.4 Congenital or acquired left ventricular outflow tract obstruction and congenital cardiomyopathies
- 3. Pulmonary hypertension due to lung disease and/or hypoxia
- 3.1 Chronic obstructive pulmonary disease
- 3.2 Interstitial lung disease
- 3.3 Mixed obstruction and restriction
- 3.4 Sleep disordered breathing
- 3.5 Chronic exposure to high altitude
- 4. Chronic thromboembolic pulmonary hypertension
- 5. Pulmonary hypertension due to unclear multifactorial mechanisms
* Adapted from Simonneau et al.2 † The classification group 1’ was created because PVOD and/or PCH share many characteristics with idiopathic PAH but are recognised to also have distinct differences.
Box 2 –
Diagrammatic representation of the key abnormal pathways targeted in treatment of pulmonary arterial hypertension and the mode of action of current and new drugs
Box 3 –
Drug treatments for idiopathic pulmonary arterial hypertension (PAH)
|
|
Class of drug
|
|
Endothelin receptor antagonists
|
PDE5 inhibitors
|
Prostanoids
|
Calcium channel blockers
|
sGC stimulators
|
Prostacyclin receptor agonists
|
|
|
Generic names
|
Bosentan, ambrisentan, macitentan
|
Sildenafil, tadalafil
|
Epoprostenol, iloprost
|
Nifedipine, amlodipine, diltiazem
|
Riociguat
|
Selexipag
|
|
Route of administration
|
Oral
|
Oral
|
Intravenous, inhaled, oral
|
Oral
|
Oral
|
Oral
|
|
Clinical improvements
|
6MWD, PVR, TCD, fewer morbidity and mortality events*
|
6MWD, TCD†
|
6MWD, TCD, haemodynamics,‡ survival,‡ quality of life‡
|
Survival, haemodynamics, functional capacity
|
6MWD, TCD, haemodynamics, functional capacity
|
Reduced death, hospitalisation and worsening of PAH; haemodynamics
|
|
Common side effects
|
Hepatotoxicity,§ peripheral oedema,¶ nasal congestion,¶ nasopharyngitis,* headache,* anaemia
|
Headache, dyspepsia, diarrhoea, myalgia, flushing, epistaxis
|
Central venous access infection or blockage,‡ cough, headache, flushing, jaw pain
|
Hypotension, peripheral oedema
|
Headache, dyspepsia, peripheral oedema, dizziness, hypotension
|
Headache, nasopharyngitis, anaemia, jaw pain, nausea
|
|
|
PDE5 = phosphodiesterase type 5. sGC = soluble guanylate cyclase. 6MWD = 6-minute walk distance. PVR = pulmonary vascular resistance. TCD = time to clinical deterioration. * Macitentan only. † Tadalafil only. ‡ Epoprostenol only. § Bosentan only. ¶ Ambrisentan only.
|